Gastrointestinal developmental disorders
Liver
Agenesis or hypoplasia of the right lobe of the liver is rare. Ectopic liver tissue may be attached to the spleen, adrenal glands, or pancreas.
Duodenal stenosis and atresia
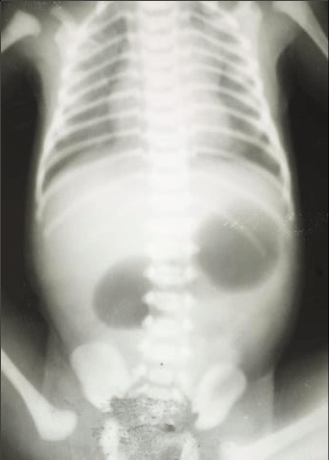
Duodenal stenosis and atresia occur due to defects in recanalization of the duodenum. They are most often seen in the region surrounding the ampulla of Vater.
Prenatally, polyhydramnios may be present.
Postnatally, typical findings include:
- Bilious vomiting, usually beginning a few hours after birth
- Abdominal distension
- Constipation
- Jaundice
A “double-bubble” sign may be seen on abdominal X-ray. About 30% of cases are associated with Down syndrome.
Many cases are also associated with:
- Annular pancreas
- Malrotation
- TEF
- Cardiac malformations (e.g., PDA and endocardial cushion defects)
- Renal defects
A few cases are inherited as an autosomal recessive trait.
Jejunoileal atresias
In contrast to duodenal atresia, jejunoileal atresias are caused by disruption of the blood supply to the small intestine. The underlying cause is a partially or completely absent mesentery, which can allow twisting of the intestine around the marginal artery, leading to ischemia and cell death. The vascular insult may also be due to local causes such as thrombosis and vasoconstriction.
Infants of mothers with the following exposures are at high risk for jejunoileal atresias:
- Ergotamine
- Excess caffeine
- Cocaine abuse
- Pseudoephedrine
- Smoking during pregnancy
It may present with:
- Failure to thrive
- Bilious vomiting
- Abdominal distension
- Jaundice
- Failure to pass meconium after birth
- Constipation
Prenatal ultrasound or MRI can be used for diagnosis. Imaging may show polyhydramnios, dilated bowel loops with air-fluid levels, hyperechoic bowel, and ascites.
Developmental anomalies of the vitelline duct or omphalomesenteric duct
The vitelline duct connects the yolk sac to the midgut. These anomalies are more common in males.
-
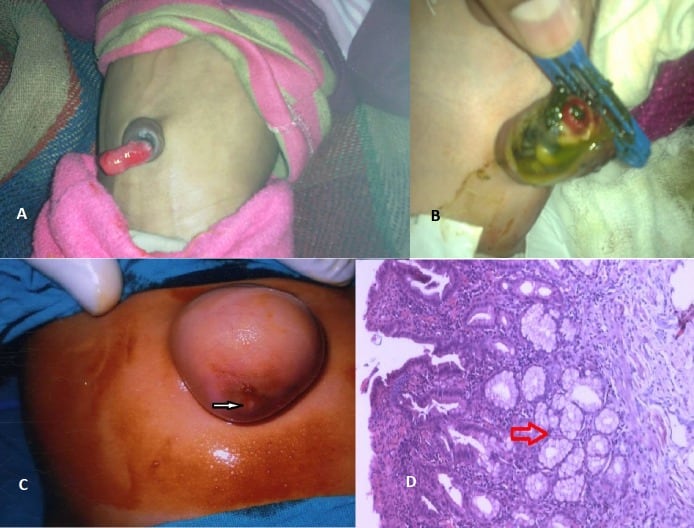
Meckel’s diverticulum: This is the most common congenital anomaly of the small intestine. It is caused by incomplete obliteration of the vitelline duct. It is a true diverticulum (contains all three layers of the ileum) and is located on the antimesenteric border of the ileum. Classically, it follows the “rule of 2s”: typically 2 feet proximal to the ileocecal valve, seen in 2% of the population, about 2 inches long, often presents around age 2 years, about 2 cm wide, 2 times more common in males, and contains 2 types of ectopic tissue (gastric and pancreatic).
Gastric mucosa can ulcerate and cause painless rectal bleeding/hematochezia or ulceration. A fibrous band connected to the tip of the diverticulum may cause intestinal obstruction. Meckel’s diverticulitis can mimic appendicitis clinically, with periumbilical and right iliac fossa pain.
Technetium 99m pertechnetate scan is positive, especially when there is a history of bleeding. Pentagastrin, glucagon, or cimetidine can be used to increase the sensitivity and specificity of the scan. SMA arteriogram can be done when the rate of bleeding is higher than 1 ml/min.
Figure 1: A- Prolapsed patent VID. B- Meconium discharge from patent VID. C- Umbilical hernia harboring Meckel’s diverticulum. Arrow shows site of attachment of Meckel’s diverticulum to the underside of skin. D- Histopathology showing ectopic gastric mucosa (Arrow).
-
Persistent vitelline duct: This occurs when the vitelline duct fails to close. It presents as a persistent draining fistula at the umbilicus.
-
Omphalomesenteric duct cyst: This occurs when the vitelline duct obliterates at both ends but remains patent in the middle. The cyst attaches to the umbilicus and/or bowel.
Omphalocele or Exomphalos
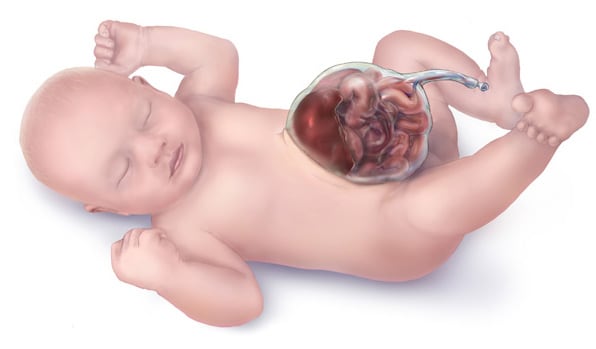
Omphalocele is a ventral abdominal wall defect caused by failure of the midgut loops to return to the abdominal cavity during fetal development. Herniation of visceral organs occurs in the midline at the base of the umbilical cord, and the contents are covered by parietal peritoneum, amnion, and Wharton jelly. The liver is a common content of an omphalocele.
Polyhydramnios or oligohydramnios and fetal ascites may be detected. Amniotic fluid AFP and acetylcholinesterase levels may be elevated.
Risk is increased in infants born to mothers who:
- Smoke or drink alcohol
- Are obese
- Take SSRIs
Omphalocele is commonly associated with:
- Chromosomal abnormalities
- Neural tube defects
- Cleft palate
- Limb defects
- Beckwith-Wiedemann syndrome
- Exstrophy of bladder
- Anal atresia
- Renal defects
- Cardiac defects
Gastroschisis
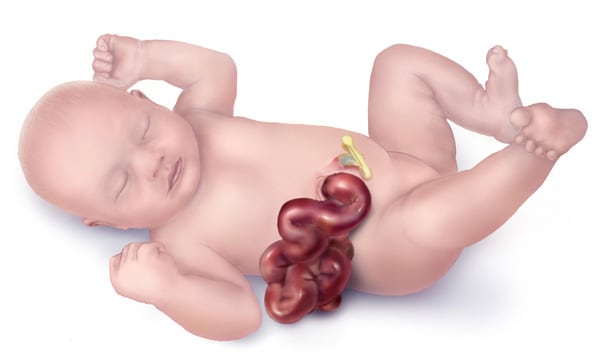
Gastroschisis is a ventral abdominal wall defect in which abdominal contents herniate through a paramedian defect, typically located to the right of the umbilicus. The bowel is not covered by a membrane.
In contrast to omphalocele, the liver never herniates in gastroschisis.
Both maternal serum and amniotic fluid AFP levels are elevated. Prenatal ultrasound shows herniated bowel and polyhydramnios. IUGR is frequently associated.
Risk is increased in infants born to:
- Teenage mothers
- Mothers who smoke or consume alcohol
GERD may develop postnatally.
Malrotation
During development, the gut undergoes a 270-degree rotation around the superior mesenteric artery (SMA) as its axis, followed by fixation in the normal anatomical position. Abnormal rotation and/or fixation leads to malrotation.
Depending on the stage at which rotation and fixation are arrested, malrotation may present with:
- A narrow base of mesentery, predisposing to midgut volvulus
- Duodenal obstruction
- Misplaced and mobile cecum
- Ladd’s bands
- Internal hernias
Malrotation is commonly associated with diaphragmatic hernia, gastroschisis, omphalocele, and intestinal atresias. It is more common in males.
Midgut volvulus is a feared complication. It presents with sudden-onset bilious vomiting, diffuse abdominal pain, and distension. If not treated immediately with surgery, it may cause bowel gangrene.
Ladd’s bands are fibrous peritoneal bands extending from the misplaced cecum and crossing over the duodenum. They may compress the duodenum and cause obstruction.
Imaging in malrotation may show signs of intestinal obstruction, with pneumatosis intestinalis in severe cases with sepsis. Upper GI series shows variable findings, including abrupt cessation of contrast, a corkscrew pattern, obstruction, etc.
Mutations in the BCL6 gene have been implicated in malrotation.
Cloaca and malformations
The terminal hindgut ends in an endoderm-lined pouch called the cloaca. The cloaca also communicates with the lower urogenital tract.
The urorectal septum (mesodermal in origin) divides the cloaca into:
- The urogenital sinus (ventral)
- The rectoanal canal (dorsal)
The rectoanal canal forms the rectum and upper anal canal, while the urorectal septum forms the perineal body.
The cloacal membrane is formed by endoderm from the cloaca and ectoderm of the opposing body wall. Normally, the cloacal membrane disintegrates. The lower anal canal is formed by fusion of ectodermal anal tubercles (proctodeum).
-
Persistent cloaca syndrome: The cloaca persists, causing the rectum, vagina, and urinary tract to open through a single opening located at the normal site of the urethra. It is often associated with tethered spinal cord.
-
Imperforate anus: This is an umbrella term for anorectal malformations presenting with absence of a normally located anal opening. The cloacal membrane fails to break down. The anorectal canal instead opens as a fistula into the perineum and connects to urinary and reproductive tract structures. It has often been associated with Down syndrome, single umbilical artery, VACTERL defects, in vitro fertilization, maternal diabetes, and intranatal exposure to thalidomide and retinoic acid. It is more common in males. VSD and PDA can be present.
Hirschsprung disease or congenital megacolon
Neurons in the submucosal and myenteric plexuses are derived from the neural crest. In Hirschsprung disease, neural crest cells fail to migrate into the colon, resulting in an aganglionic segment.
Because the aganglionic segment cannot relax, it remains tonically contracted and causes functional obstruction. The proximal colon dilates, which is why it is called congenital megacolon. The sigmoid colon is the most commonly affected segment.
It presents with:
- Failure to pass meconium at birth
- Poor weight gain
- Slow growth
- Constipation
- Bowel obstruction
- Vomiting
Complications include enterocolitis and toxic megacolon.
Rectal examination shows a tight anal sphincter and explosive discharge of stool and gas.
RET, SEMA3, NRG1, and EDNRB genes have been implicated in the pathogenesis of Hirschsprung disease. It is associated with Down syndrome, cartilage hair hypoplasia syndrome, MEN type 2, etc.
Imaging shows a classic narrowed distal colon with proximal dilation and a transition zone in between. Rectal contrast is retained for more than 24 hours following a barium enema in Hirschsprung disease.
Anorectal manometry (not done commonly) shows lack of relaxation reflex of the internal sphincter after rectal distension.
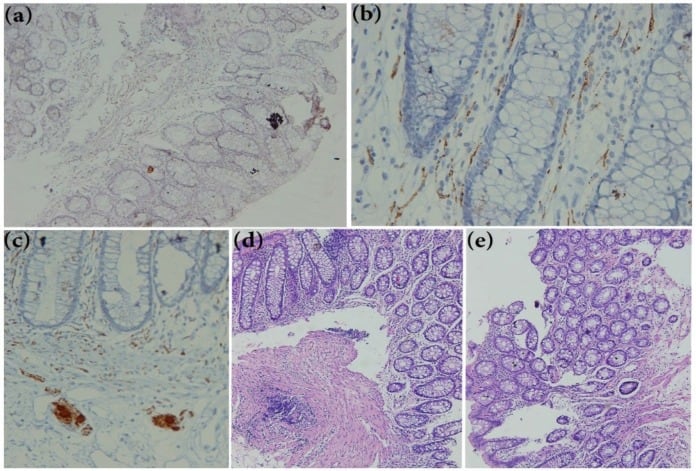
The gold standard for diagnosis is a full-thickness rectal biopsy showing absent ganglion cells. Suction biopsy can also be done, though it is less sensitive. H and E, acetylcholinesterase staining (showing hypertrophied nerve trunks in the bowel wall), or immunohistochemical staining with calretinin are used.
Rectal biopsy sections of suspicion patient for Hirschsprung disease (HD). (a) Showing negative reaction after calretinin immunohistochemistry staining (×250). (b) Calretinin positive immunohistochemistry on a normal rectal biopsy sections extended to the lamina propria easily detectable at ×400 magnification in submucosae. © Calretinin positive Immunoreactivity in Ganglion cells of submucosa (×400). (d, e) H&E staining rectal biopsy of (d) normal tissue and (e) HD patient