Hemolytic anemias
Anemia caused by hemolysis (breakdown of RBCs) is called hemolytic anemia. In hemolysis, RBCs are destroyed earlier than their normal life span of about 120 days, which lowers the RBC count and leads to anemia.
Broadly, hemolytic anemia is classified as intravascular or extravascular:
- Intravascular hemolysis: RBCs are destroyed within the circulation. Hemoglobin is released into the plasma and may appear in urine, causing hemoglobinuria.
- Extravascular hemolysis: RBCs are removed and broken down by macrophages of the reticuloendothelial system (mainly in the spleen and liver). Because hemoglobin is processed inside macrophages, plasma hemoglobin doesn’t rise much, and hemoglobinuria is absent.
Causes of intravascular and extravascular hemolytic anemia
Intravascular hemolysis
- AIHA
- Drug induced autoimmune hemolytic anemia e.g. meropenem, iron dextran, ceftriaxone, piperacillin
- Mechanical trauma like artificial heart valves, calcified aortic valve
- G6PD deficiency
- TTP, HUS
- DIC, HELLP syndrome
- PNH
- Mismatched blood transfusion
- Toxins in malaria (blackwater fever), clostridial toxins, sepsis, snake venom
Extravascular hemolysis
- AIHA
- Hereditary spherocytosis
- Hereditary elliptocytosis
- Pyruvate kinase deficiency
- Sickle cell anemia
- Thalassemias
Normally, haptoglobin in the plasma binds free hemoglobin. In severe intravascular hemolysis, haptoglobin levels decrease and may become absent. This happens because the haptoglobin-hemoglobin complex is cleared by the reticuloendothelial system faster than the liver can increase haptoglobin synthesis.
Comparison between intravascular and extravascular hemolytic anemia
| Intravascular hemolysis | Extravascular hemolysis |
| Haptoglobin decreased or absent | Haptoglobin levels are normal or mildly decreased |
| Hemoglobinuria may be present | Hemoglobinuria absent |
| Urine hemosiderin increases | Urine hemosiderin absent |
| LDH elevated | LDH elevated |
| Helmet cells may be seen | Spherocytes may be seen |
| Total bilirubin may increase Serum unconjugated bilirubin may be normal or may increase | Total bilirubin increases Serum unconjugated bilirubin increases |
| Conjugated bilirubin is normal | Conjugated bilirubin usually normal |
| Reticulocytosis seen | Reticulocytosis seen |
-
Auto-immune hemolytic anemia or AIHA: AIHA is an autoimmune disorder in which autoantibodies target RBCs and cause hemolysis. It can cause intravascular or extravascular hemolysis.
AIHA may be:
- Primary (idiopathic)
- Secondary (acquired)
Secondary causes include autoimmune diseases (such as lupus), chronic lymphocytic leukemia, non-Hodgkin’s lymphoma and other blood cancers, Epstein-Barr virus, Cytomegalovirus, Mycoplasma pneumonia, Hepatitis viruses and HIV. Some drugs can also induce autoantibody production against RBCs.
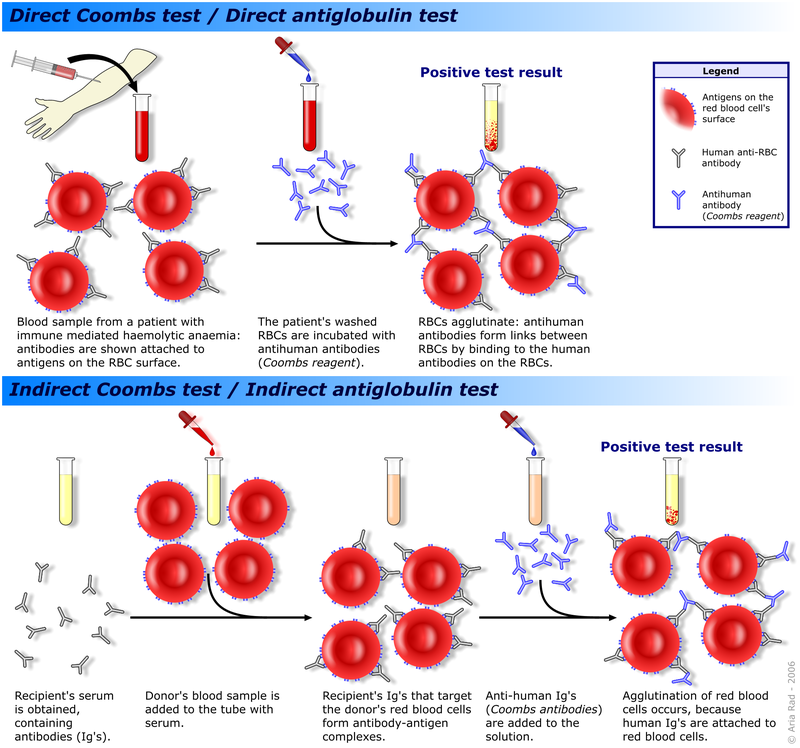
In addition to symptoms of anemia, severe cases may show jaundice and splenomegaly. DAT and the indirect antibody test are positive in AIHA.
Acquired autoimmune hemolytic anemia occurs in different forms, including warm antibody hemolytic anemia (WAIHA) and cold antibody hemolytic anemia (CAIHA).
- In WAIHA, autoantibodies react optimally with human RBCs at 37ºC.
- In CAIHA, hemolysis is mediated by cold autoantibodies that bind RBCs at temperatures below 37ºC.
WAIHA is the more common form and is associated with IgG1 or IgG3 antibodies. It is seen in CLL, non-Hodgkin’s lymphoma, solid tumors, SLE, viral infections, drugs like cephalosporins and piperacillin; previous transfusions or transplantation.
CAIHA is caused by antibodies that react optimally at 4°C and includes cases of cold agglutinin syndrome (CAS) and paroxysmal cold hemoglobinuria (PCH).
CAS characteristically occurs in middle-aged or elderly persons, often with signs and symptoms worsened by cold, such as livedo reticularis, Raynaud disease, acrocyanosis, and cutaneous necrosis. CAS is associated with IgM and may be primary (a nonmalignant clonal B cell disorder) or secondary to non-Hodgkin’s lymphoma, CLL, Mycoplasma pneumoniae or infectious mononucleosis (Epstein-Barr virus).
The diagnosis of CAS is suggested by autoagglutination of the patient’s blood at room temperature that is worsened at 4°C and reverses at 37°C. Patients with CAS have a positive DAT with anti-C3 or anti-C3d, but are negative with anti-IgG reagents.
PCH is more common in children and follows a viral illness or vaccination. It presents with intravascular hemolysis with hemoglobinuria and jaundice, along with abdominal pain, cutaneous and mucosal pallor, fever, chills, and chronic headache. Symptoms are typically preceded by 1-2 weeks of a respiratory tract infection. Renal failure may occur.
PCH is caused by a cold agglutinin called the Donath-Landsteiner antibody, a polyclonal IgG biphasic antibody that binds RBCs at cold temperatures and produces complement mediated intravascular hemolysis at normal body temperature.
Rarely, some patients have mixed AIHA with both warm and cold antibodies. Treatment of AIHA depends on severity and type. Corticosteroids, rituximab, avoiding cold exposure, cyclosporine, splenectomy and blood transfusions are used as therapy.
-
Drug-induced hemolytic anemia or DIHA: DIHA is caused by drug-related antibodies that lead to RBC breakdown. A key point is that DIHA can be differentiated from AIHA because it typically goes into remission after discontinuation of the offending drug.
Following mechanisms are seen:
i) Drug binds to RBC membrane proteins inducing typically IgG antibody production against the complex. Such antibody coated RBCs are removed by macrophages. Classic example is Penicillin, which binds covalently to RBC proteins. Diagnosed by positive DAT and negative antibody elution tests. Indirect antiglobulin test may be positive or negative. Drug dependent antibodies seen.
ii) Combination of membrane plus drug can create an immunogen, stimulating the production of IgM or IgG antibodies and often activate complement, leading to acute intravascular lysis and sometimes renal failure. Seen with ceftriaxone, piperacillin, NSAIDS, quinine, probenecid etc. Diagnosed by positive DAT and negative antibody elution tests. Indirect antiglobulin test may be positive or negative. Drug dependent antibodies seen.
iii) Modification of the immune system so that RBC auto-antibodies are produced e.g. Methyl dopa, fludarabine. Drug independent antibodies seen. Diagnosed by positive DAT, indirect antiglobulin and antibody elution tests.
iv) Some drugs modify the RBC membrane so that proteins such as albumin, IgG, C3, fibrinogen etc, become non-immunologically adsorbed onto the RBC membrane, activating an immune response. Drug independent antibodies seen. Diagnosed by positive DAT, indirect antiglobulin and antibody elution tests.
Common drugs causing DIHA
- Methyldopa
- Penicillin
- Cephalosporins like ceftriaxone, cefotetan
- Piperacillin
- NSAIDS
- Quinine
- Hydrochlorothiazide
- Rifampicin
- Mefloquine
- Fludarabine
- Probenecid
- Beta lactamase inhibitors
-
Paroxysmal nocturnal hemoglobinuria or PNH or Marchiafava-Micheli Syndrome:
Paroxysmal nocturnal hemoglobinuria is a condition affecting RBCs, WBCs and platelets. It is characterized by sudden, recurring episodes of intravascular hemolysis and hemoglobinuria, which may be triggered by stressors such as infections or physical exertion.
PNH is caused by acquired somatic mutations in the PIGA gene in hematopoietic stem cells. The PIGA gene codes for phosphatidylinositol glycan class A, a protein involved in producing the GPI anchor, which “anchors” proteins to the cell membrane. Important GPI-anchored proteins include CD55 (decay accelerating factor or DAF) and CD59. Absence of these proteins makes RBCs more susceptible to complement mediated lysis.
In many cases, hemoglobinuria is noticed in the morning as brown or cola colored urine that has accumulated in the bladder overnight. Other findings include abdominal vein thrombosis, hemorrhage, pancytopenia, thrombocytopenia. Budd-Chiari syndrome may be seen. Patients with PNH are prone to AML. It is a diagnosis of exclusion.
Flow cytometry can detect abnormal cells lacking GPI anchored proteins like CD55 and CD59. Eculizumab and Ravulizumab can be used for therapy; they act by blocking complement mediated lysis. Bone marrow transplantation is curative. Folate and/or iron supplementation may be needed. Ham’s test and sucrose hemolysis test can also be used to detect hemolysis in PNH.