Tricarboxylic acid cycle
TCA (Tricarboxylic acid cycle) or Krebs cycle or citric acid cycle
The TCA cycle is the final common pathway where the oxidative metabolism of carbohydrates, fats, and amino acids converges. Their carbon skeletons are ultimately converted to CO2. The cycle occurs entirely in the mitochondria.
The TCA cycle also supplies intermediates for many synthetic reactions, such as:
- Glucose formation from the carbon skeletons of some amino acids
- Providing building blocks for some amino acids and heme
Oxidative decarboxylation of pyruvate: Pyruvate, the end product of aerobic glycolysis, must be transported into the mitochondria before it can enter the TCA cycle. A specific pyruvate transporter helps it cross the inner mitochondrial membrane. Once in the mitochondrial matrix, pyruvate is converted to acetyl CoA by the multienzyme pyruvate dehydrogenase (PDH) complex.
The PDH complex has two tightly bound regulatory enzymes:
- PDH kinase
- PDH phosphatase
There are five coenzymes for PDH: thiamine pyrophosphate (TPP), lipoic acid, CoA, FAD, and NAD. These act as carriers or oxidants.
Thiamine /Niacin Deficiency: If PDH can’t function, ATP supply via the TCA cycle is limited. The CNS is affected more commonly (e.g., Wernicke-Korsakoff psychosis in alcoholics results from thiamine deficiency).
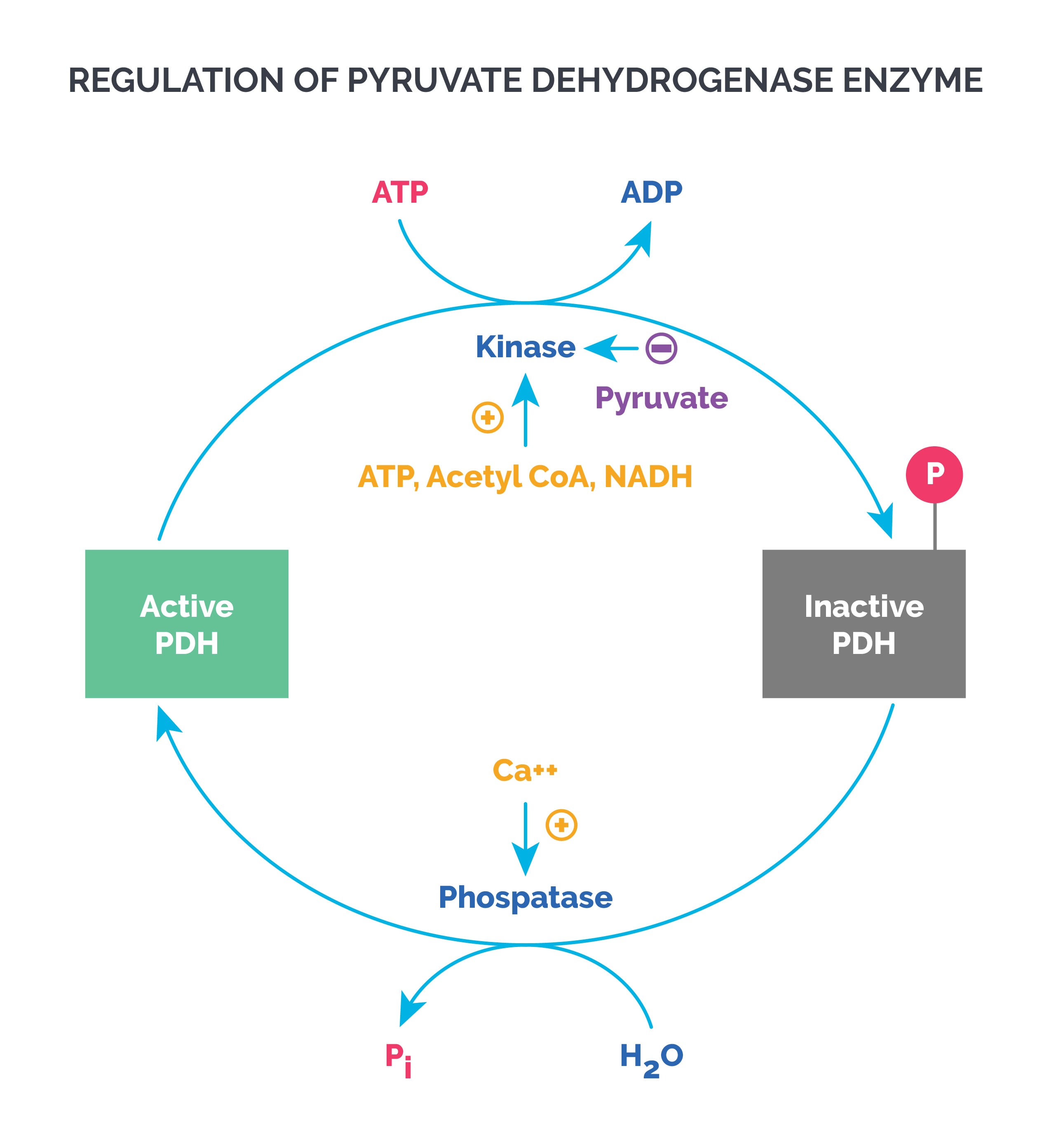
Regulation of PDH: PDH kinase phosphorylates and inactivates PDH, while PDH phosphatase activates PDH.
- PDH kinase is allosterically activated by ATP, acetyl CoA, and NADH (therefore inhibiting PDH).
- Pyruvate is a potent inhibitor of pyruvate kinase (activates PDH).
- Calcium activates pyruvate phosphatase (activates PDH).
Remember that TCA cycle enzymes, just like glycolysis, are active when dephosphorylated.
PDH Deficiency: Causes congenital lactic acidosis. CNS effects include neurodegeneration, muscle spasticity, and early neonatal death. It is X-linked dominant, so it affects both males and females because the enzyme is vital to life. Leigh’s syndrome (subacute necrotizing encephalomyelopathy) is due to defects in mitochondrial ATP production, mainly from mutations in the PDH complex, ETC, or ATP synthase enzyme. Both nuclear and mitochondrial DNA may be involved.
Arsenic poisoning: In addition to interfering with glycolysis, arsenic inhibits enzymes that require lipoic acid as a coenzyme, such as PDH, alpha ketoglutarate dehydrogenase, and branched chain alpha keto acid dehydrogenase. Arsenite (trivalent arsenic) forms a stable complex with thiol (-SH) groups in lipoic acid and inhibits the enzyme. Pyruvate and lactate accumulate, affecting the brain more commonly and potentially causing death.
The TCA cycle occurs in the following steps:
-
Oxaloacetate combines with Acetyl CoA to form Citrate (a tricarboxylic acid) by citrate synthase. Citrate inhibits PFK 1 of glycolysis while activating acetyl CoA carboxylase of fatty acid synthesis.
-
Citrate is isomerized to isocitrate by enzyme aconitase, an Fe-S protein. Aconitase is inhibited by fluoroacetate in rat poison.
-
Isocitrate is converted to alpha ketoglutarate by isocitrate dehydrogenase, yielding 3 NADH. This is a rate-limiting step of the TCA cycle. The enzyme is allosterically activated by ADP and calcium and is inhibited by ATP and NADH.
-
Alpha keto glutarate is converted to succinyl CoA by the enzyme complex alpha ketoglutarate dehydrogenase. It has 5 coenzymes: TPP, CoA, lipoic acid, FAD, and NAD. The enzyme is inhibited by succinyl CoA and NADH and activated by calcium. Transamination of amino acids (glutamate) yields alpha ketoglutarate.
-
Succinyl CoA is cleaved to succinate by succinate thiokinase (also called succinyl CoA synthase). This is a substrate-level phosphorylation that yields 1 GTP. Succinyl CoA is also produced from propionyl CoA from metabolism of odd chain fatty acids and from some amino acids.
-
Succinate is oxidized to fumarate by succinate dehydrogenase, forming FADH2. This enzyme functions as Complex II of the ETC.
-
Fumarate is hydrated to malate by fumarate hydratase. Fumarate is also produced in the urea cycle, purine synthesis, and during the catabolism of amino acids phenylalanine and tyrosine.
-
Malate is oxidized to oxaloacetate by malate dehydrogenase. NADH is also produced. Oxaloacetate is also produced by the transamination of aspartic acid.
Energy yield in TCA cycle: For each molecule of acetyl CoA that enters the cycle, 3 NADH, 1 FADH2, and 1 GTP are produced.
- 1 NADH yields 3 ATP
- 1 FADH2 yields 2 ATP
- 1 GTP yields 1 ATP
So, in total, 12 ATP are produced by the TCA cycle per molecule of acetyl CoA oxidized. Note that 1 molecule of glucose yields 2 molecules of acetyl CoA, so the TCA cycle produces 24 ATP per glucose.
Depending on which shuttle is used by NADH to re-enter the mitochondrial matrix:
- 2 ATP are produced using the glycerol phosphate shuttle
- 3 ATP are produced using the malate-aspartate shuttle