Glycogen Metabolism
Main stores of glycogen are the liver and skeletal muscle.
- Liver glycogen helps maintain blood glucose levels.
- Muscle glycogen is used locally as fuel to generate ATP during muscle contraction and can’t be used to raise blood glucose levels.
Glycogen is a branched-chain polysaccharide made from alpha D glucose.
Glycogen synthesis or glycogenosis: It occurs in the cytosol and requires ATP and UTP.
- UDP glucose is the activated component essential for glycogen formation.
- Glucose 1 phosphate is converted to UDP glucose by UDP glucose pyrophosphorylase.
- Glycogen synthase elongates the chain by adding alpha 1,4 bonds.
- If there’s no pre-existing glycogen fragment to serve as a primer, glycogenin can be used to start glycogen synthesis.
- Branch points are made by the branching enzyme (transglucosidase or 4:6 transferase), which breaks the glycogen molecule at C4 and then attaches it to C6.
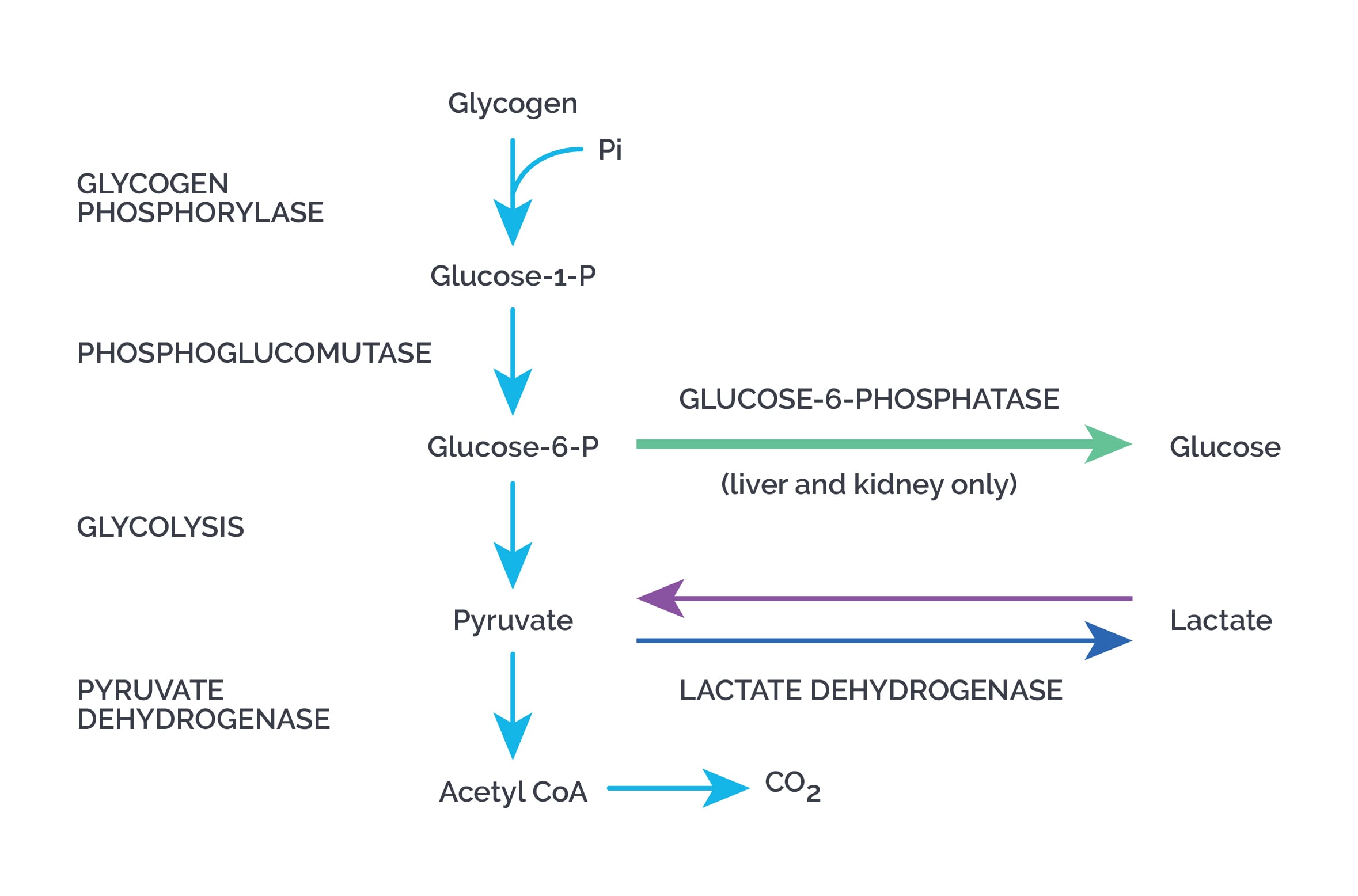
Degradation of glycogen or glycogenolysis:
- Alpha 1,4 bonds are broken to yield glucose 1 phosphate by glycogen phosphorylase until four glucose units remain on each chain before the branch point, forming limit dextrins.
- Glycogen phosphorylase requires pyridoxal phosphate.
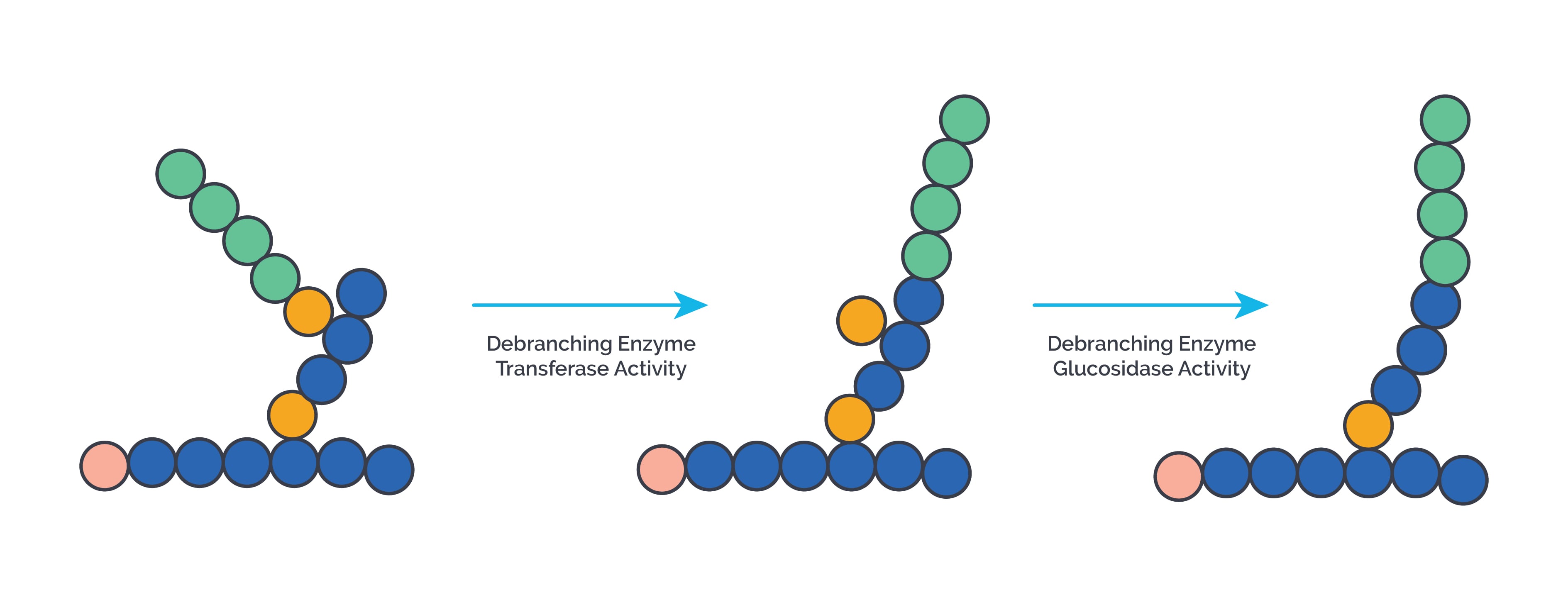
- Branches are removed by the debranching enzyme, which acts as 4,4 glucan transferase, 1,4 glucan transferase, and 1,6 glucosidase.
- Alpha 1,6 bonds are broken to release free glucose.
Glucose 1 phosphate is converted to glucose 6 phosphate by phosphoglucomutase.
- In the liver, glucose 6 phosphate is transported into the ER by a translocase and then converted to glucose by glucose 6 phosphatase.
- A small amount of glycogen is degraded by a lysosomal enzyme, alpha 1,4 glucosidase (acid maltase). Deficiency causes Pompe’s disease.
Regulation of glycogen metabolism: Glycogen formation occurs when the body is well fed, whereas glycogen breakdown happens during fasting and in skeletal muscle during exercise.
- Regulatory enzymes for glycogen formation are inactive when phosphorylated and active when dephosphorylated (same as for glycolysis).
- For glycogenolysis, it is exactly the opposite.
Glucagon acting on its receptor in the liver and epinephrine acting on the beta receptor on muscle and liver activate adenylyl cyclase, increasing cAMP levels. cAMP activates protein kinase A, which activates phosphorylase kinase. This leads to phosphorylation and activation of glycogen phosphorylase, which breaks down glycogen. At the same time, activated protein kinase A phosphorylates and inactivates glycogen synthase, inhibiting glycogen synthesis.
Insulin activates a phosphodiesterase that breaks down cAMP, ending the action of glucagon and epinephrine.
Regulation by calcium: Epinephrine via the alpha 1 receptor on the liver and neural stimulation of muscle causes calcium release into the cytosol, which activates phosphorylase kinase. This ultimately stimulates glycogen breakdown and inhibits glycogen synthesis.
Allosteric regulation of glycogen metabolism
| Site | Glycogen phosphorylase | Glycogen synthase |
| Liver | Inhibited by glucose, ATP and glucose 6 phosphate | Activated by glucose 6 phosphate |
| Muscle | Inhibited by ATP and glucose 6 phosphate; Activated by calcium and AMP | Activated by glucose 6 phosphate |
Glycogen storage disorders: All glycogen storage disorders are inherited as autosomal recessive except some cases of type IV.
-
Type I or Von-Gierke disease: It is of two types- Ia due to the deficiency of glucose-6-phosphatase, and Ib due to the deficiency of glucose-6-phosphate translocase. It is more prevalent in Ashkenazi Jews. Mutations are seen in G6PC and SLC37A4 genes. Symptoms typically start at age 3 or 4 months. It presents with severe fasting hypoglycemia, fatty liver, hepatomegaly, renomegaly, seizures, growth retardation and delayed puberty. Laboratory findings include lactacidemia, hyperlipidemia, xanthomas, diarrhea, osteoporosis, gout, pulmonary hypertension, PCOS and hyperuricemia. Structurally normal glycogen is stored in excess. Type Ib has in addition, neutropenia, oral ulcers, gingivitis, recurrent infections and increased risk of IBD. Treatment is with nocturnal glucose infusions, frequent feedings of uncooked cornstarch, and management of associated conditions like hyperuricemia etc.
-
Type II or Pompe disease: It is due to deficiency of lysosomal alpha 1-4 glucosidase (acid maltase) deficiency. It can present in infancy or late adulthood. Mutations in the GAA gene are seen. Excess glycogen accumulates in lysosomes as it cannot be degraded. It presents with massive cardiomegaly, hypertrophic cardiomyopathy, heart failure, large tongue, hepatomegaly, hypotonia, skeletal muscle weakness, breathing difficulties and respiratory failure . There is no hypoglycemia. Glycogen structure is normal. Enzyme replacement therapy with intravenous recombinant human alpha glucosidase is done.
-
Type III or Cori disease: It is due to deficiency of debranching enzyme (4:4 transferase or 1:6 glucosidase). Mutations in AGL genes are seen. It presents with fasting hypoglycemia, hepatomegaly, muscle weakness, short stature, hyperlipidemia, elevated ketone bodies in blood and urine, nosebleeds, recurrent infections, cardiac hypertrophy, liver cirrhosis and hepatocellular carcinoma may occur in adulthood. Abnormal glycogen with four or one glucose residues at branch points called limit dextrins, are deposited. Treatment is with frequent feeding of a high protein diet , cornstarch and avoiding fasting.
-
Type IV or Anderson disease: It results from deficiency of the branching enzyme. Anderson disease typically begins in infancy and presents with failure to thrive, enlarged liver and spleen (hepatosplenomegaly), and in many cases, progressive liver cirrhosis and liver failure, muscle weakness, cardiomyopathy and joint contractures. Mutations in the GBE1 gene are seen. Biopsy shows PAS-positive deposits of abnormal glycogen called polyglucosan. Treatment is with high-protein diet, liver transplantation and management of associated conditions.
-
Type V or McArdle disease: It is due to deficiency of skeletal muscle glycogen phosphorylase or myophosphorylase. Mutations in the PYGM gene are seen. Liver is not involved. It presents with muscle weakness and cramping after exercise. The blood lactate level fails to increase even during strenuous exercise. Laboratory findings include myoglobinuria, myobloinemia and increased creatine kinase. Glycogen structure is normal. Management includes intake of simple carbs before exercising and avoiding strenuous activities.
-
Type VI or Hers disease: It is due to deficiency of liver phosphorylase. It presents with milder symptoms, mild fasting hypoglycemia, hypotonia, mild muscle weakness, slow growth, hepatomegaly which reverts in adulthood. Mutations in the PYGL gene are seen. Treatment is by avoidance of fasting.