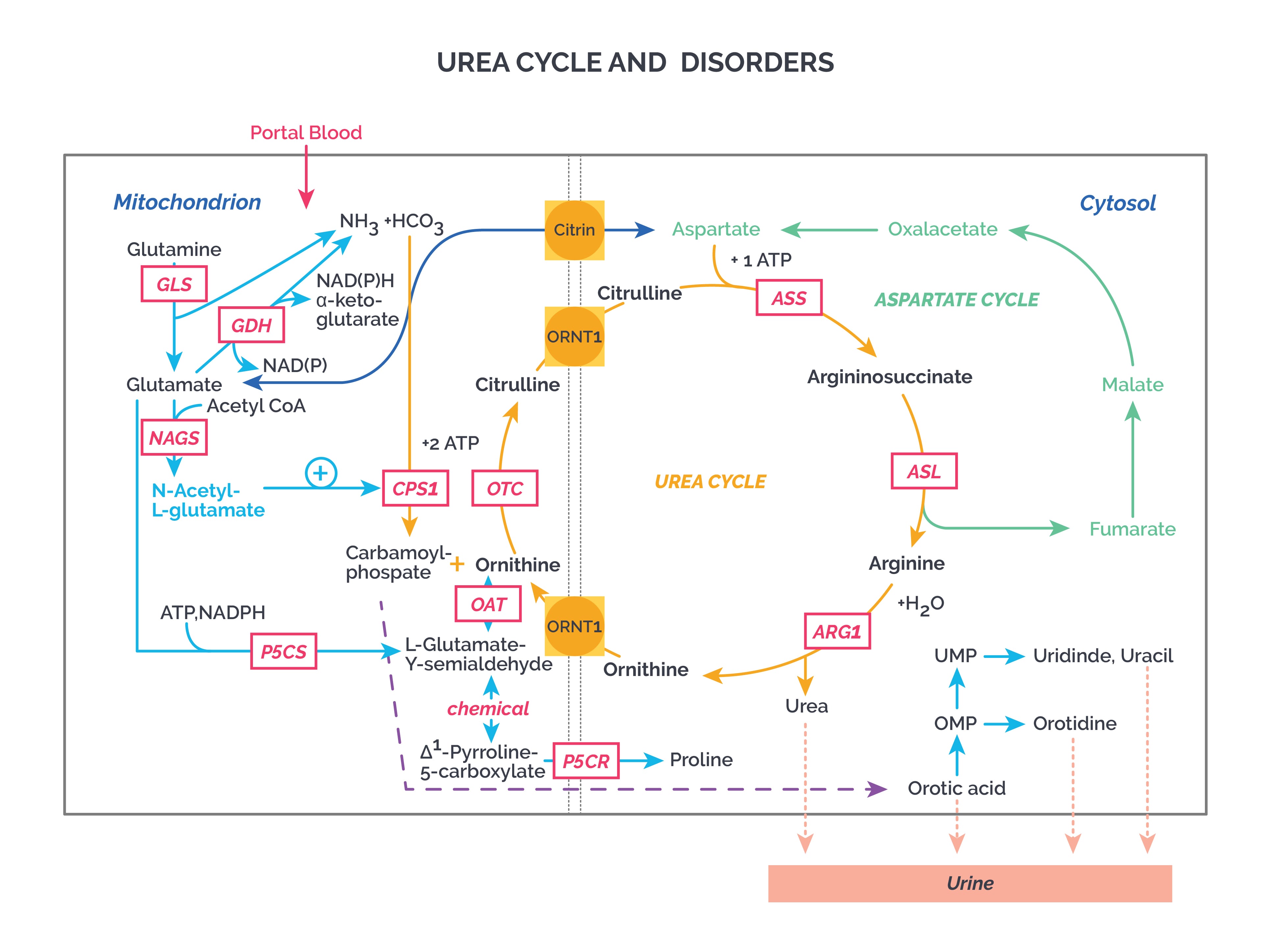
Urea cycle disorders
Urea cycle disorders (UCDs) are genetic mutations that cause defects in handling the extra nitrogen produced when protein and other nitrogen-containing molecules are broken down. This leads to accumulation of ammonia and other precursor metabolites, often within the first few days of life. Infants with a UCD may look normal at first, but can rapidly develop cerebral edema and related neurologic signs. In milder (partial) urea cycle enzyme deficiencies, ammonia accumulation may be triggered by illness or stress at almost any age.
Urea cycle: Carbamoyl phosphate synthetase (CPS) and ornithine transcarbamylase I are present only in the liver and intestines. CPS 1 is the rate-limiting enzyme. Valproic acid inhibits the urea cycle.
Signs and symptoms of UCDs are as follows:
- Normal appearance at birth
- Irritability progressing to somnolence, lethargy, then coma
- Loss of thermoregulation (hypothermia)
- Feeding disruption (increases catabolism)
- Neurologic posturing (from cerebral edema)
- Seizures, dementia
- Hyperventilation and then hypoventilation
- Vomiting
- Behavior changes
All UCDs are autosomal recessive except OTC deficiency, which is X-linked. Along with CPS I and NAGS deficiency, OTC deficiency is among the most severe urea cycle disorders. Following are the UCDs:
N-acetylglutamate synthase deficiency (NAGS): NAG is a cofactor for CPS 1. In NAGS deficiency, this cofactor cannot be formed. Severe disease. Hyperammonemia.
Carbamoyl phosphate I synthase (CPS I) deficiency: Severe disease. Hyperammonemia.
Ornithine transcarbamylase (OTC) deficiency: Severe disease. More common in male patients. Hyperammonemia.
Argininosuccinate synthase deficiency (ASSD or citrullinemia I): Hyperammonemia. Less severe disease. Elevated citrulline levels.
Citrin deficiency (citrullinemia II): Citrin is an aspartate-glutamate transporter across the mitochondrial membrane. Hyperammonemia, intrahepatic cholestasis, neurological findings, jaundice, fatty liver, elevated citrulline, hyperlipidemia. Japanese or Asian ancestry.
Argininosuccinate lyase deficiency (argininosuccinic aciduria): Hyperammonemia, hepatomegaly, elevated transaminases, argininosuccinic acid elevated in blood and urine, cirrhosis, trichorrhexis nodosa (node-like fragile hair).
Arginase deficiency (hyperargininemia): Progressive spasticity of lower limbs, seizures, growth retardation, anorexia, vomiting, irritability, milder hyperammonemia, elevated arginine in blood.
Ornithine translocase deficiency (HHH syndrome): Also known as hyperornithinemia, hyperammonemia, and homocitrullinuria syndrome. Orotic acid present in urine. Reduced transport of ornithine into mitochondria. Clinical features like arginase deficiency.
Tip: Remember that NAGS, OTC, and CPS I deficiency can also cause elevated orotic acid in urine. Similarly, orotic aciduria caused by a UMPS gene defect in the pyrimidine biosynthesis pathway presents with megaloblastic anemia plus elevated orotic acid in urine.