



Benign and malignant growths of the colon
Polyps are growths that protrude into the lumen of the gastrointestinal tract. Colorectal polyps may be:
- Sessile (flat, no stalk)
- Pedunculated (attached by a fibrovascular stalk)
- Neoplastic or non-neoplastic
More than 95% of colon adenocarcinomas originate from polyps. Most arise from adenomatous polyps, which develop from mucus cells in the colonic epithelium.
Risk factors associated with a higher risk of adenomatous polyps include smoking, obesity, a low-fiber diet, red meat intake, and low calcium intake. Statins and NSAIDs are associated with a lower risk.
Colon polyps are usually asymptomatic. When symptoms occur, they may include hematochezia, abdominal pain, a change in bowel habits, or diarrhea (classically with villous adenoma).
Types of polyps
| Adenomatous polyps* | Tubular: Branched, tubular glands; most common type of adenomatous polyp; pedunculated; minimal chance of carcinoma; Villous: Finger-like projections on histology; mostly sessile; diarrhea, hypokalemia; higher risk of carcinoma; Tubulovillous: Mixed pattern; intermediate risk of carcinoma |
| Serrated polyps** | Sessile: Common, seen in proximal colon, low malignant potential, microsatellite instability seen, hypermethylation of MLH1; Traditional: Uncommon, seen in distal colon, high malignant potential, BRAF gene mutations, |
| Non-neoplastic | Hyperplastic: Most common colorectal polyp, sessile, seen in distal colon and rectum, low malignant potential, typically no dysplasia; Juvenile: Hamartomas, not premalignant, common in childhood, seen in rectosigmoid area of colon, composed of connective tissue and hypertrophied epithelium, pedunculated, cherry red, may cause bleeding, intussusception, obstruction; Inflammatory or pseudopolyps: Seen in IBD, composed of regenerating mucosa and inflammatory infiltrate; |
*ultimately, risk of progression to colon cancer is dependent on degree of dysplasia
**histology shows serrated, papillary infoldings into the crypts
Villous adenoma: low (×400, 3a) and high (×2000, 3b) power magnification views of a hematoxilin-eosin stain. The histological appearance is of long finger-like projections.
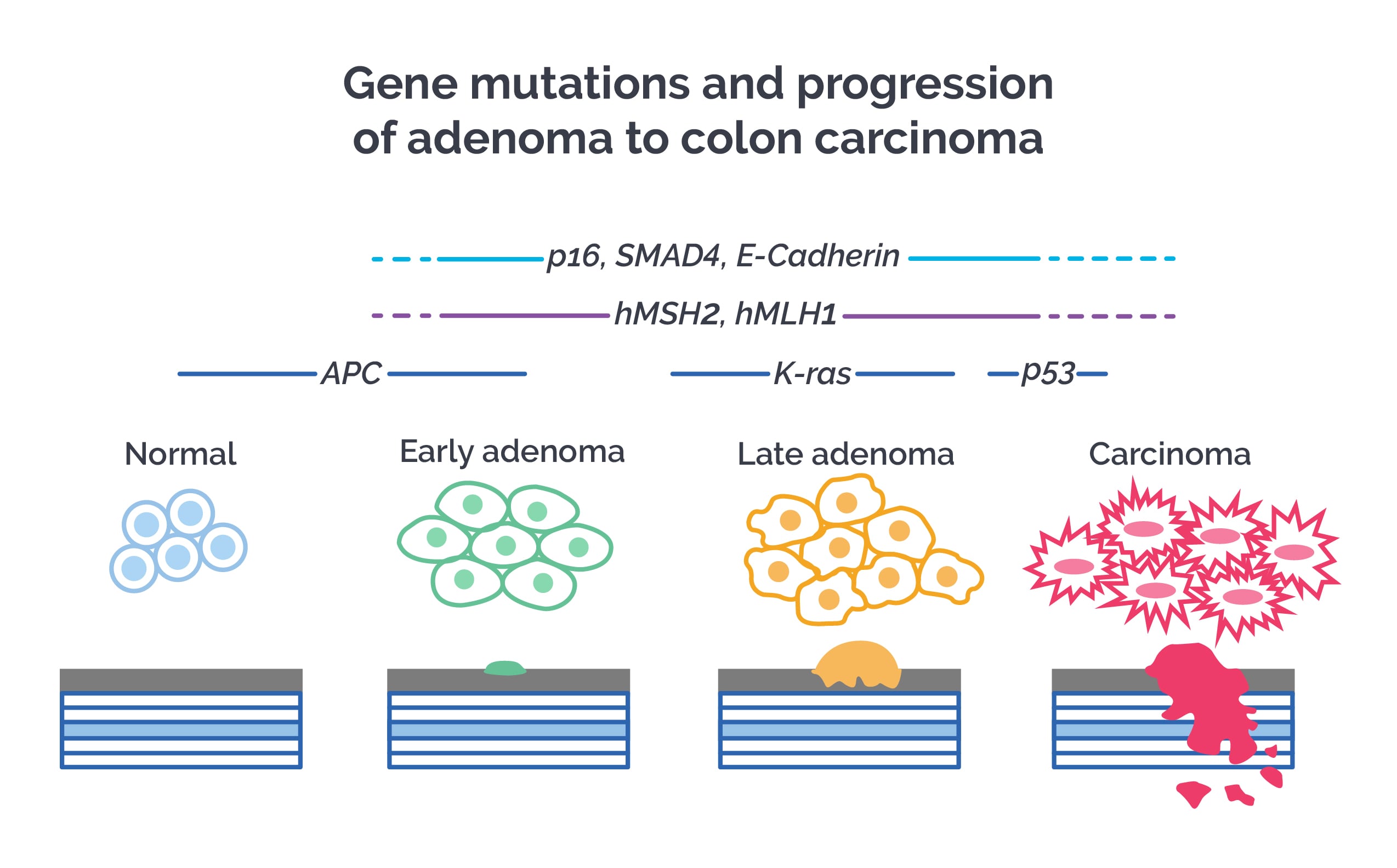
Adenomas accumulate mutations in critical proto-oncogenes, tumor suppressor genes, and DNA repair genes. Over a period of years, these changes can lead to colon carcinoma. Genetic susceptibility and environmental factors both contribute to colon carcinoma.
At the molecular level, pathogenesis involves events such as mutations, loss of heterozygosity, epigenetic silencing of gene transcription by DNA hypermethylation, gene amplification, upregulation of transcription, and translocations.
Colonoscopic view of sessile (1a) and pedunculated (1b) polyps.
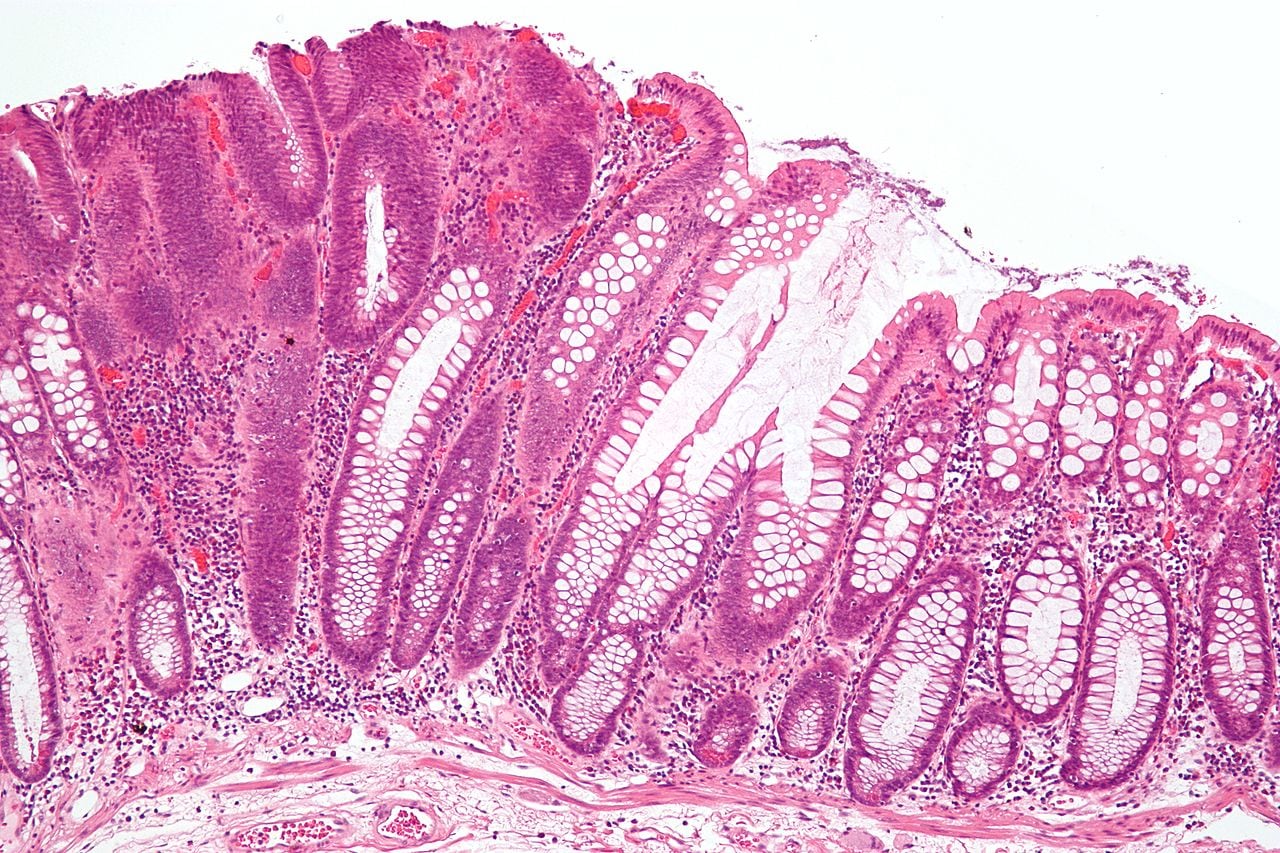
Intermediate micrograph of a colorectal tubular adenoma without high grade dysplasia. H&E stain. The lesional tissue, i.e. dysplastic epithelium, is seen on the left of the image and characterized by: Nuclear hyperchromatism (i.e. dark purple nuclei) - that extends to the surface, Nuclear crowding (i.e. nuclei are bunched-up), Elliptical/cigar-shaped nuclei and loss of goblet cells/reduction in the number of goblet cells. Normal colonic type epithelium is seen on the right of the image and characterized by small round nuclei and abundant goblet cells.
The adenoma-to-carcinoma progression begins with dysplastic foci called aberrant crypt foci, which show mutations in APC, K-ras, and TP53 genes.
- The earliest change is mutation or loss of the APC gene.
- This is followed by K-ras mutations.
- TP53 mutations occur later and mark the transition from adenoma to carcinoma.
Colon cancer developing from ulcerative colitis shows early TP53 mutations. Carcinoma confined to the muscularis mucosa does not metastasize, but once that barrier is breached, prognosis is bad.
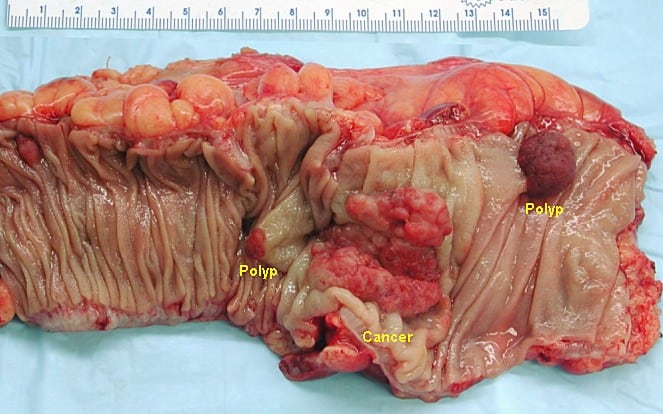
Gross appearance of a colectomy specimen containing two adenomatous polyps (the brownish oval tumors above the labels, attached to the normal beige lining by a stalk) and one invasive colorectal carcinoma (the crater-like, reddish, irregularly shaped tumor located above the label)
Some inherited syndromes associated with colon polyps are as follows -
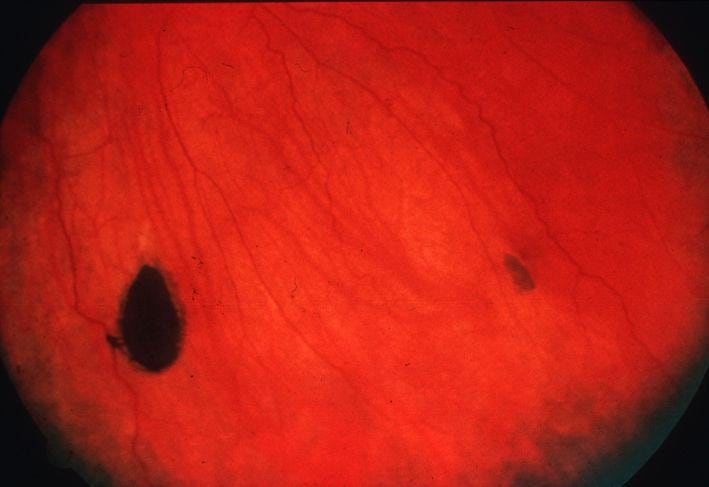
Familial adenomatous polyposis (FAP): It is an autosomal dominant disease caused by mutation in APC gene on chromosome 5q. It starts in puberty and is characterized by thousands of adenomatous polyps in the colon and rectum with 100% chance of colon cancer by the age of 40 years. Most APC mutations result in the formation of a stop codon. In less severe mutations, fewer polyps are seen and colon cancer develops later in life. Gastric, duodenal and peri-ampullary polyps (high malignant potential) are seen. Extra-intestinal manifestations include bilateral, congenital hypertrophy of the retinal pigment epithelium, osteomas of the skull, tibia and mandible, impacted or supernumerary teeth and desmoid tumors of the retroperitoneum and abdominal wall. There is increased risk of developing cancers of the thyroid (papillary carcinoma), adrenals and biliary tree.
Colonic mucosa carpeted with adenomatous polyps in a patient with familial adenomatous polyposis.
First-degree relatives of FAP patients should be screened for FAP. Initial screening should begin at age 10-12 years, followed by sigmoidoscopy (flexible proctosigmoidoscopy) every year until age 35 years. If no abnormalities are detected, screening can be done every 3 years after that. Upper gastro-intestinal endoscopy should be performed every 1-3 years after detection of colonic polyps. Genetic testing should be done to look for mutations.
Treatment of FAP is by total proctocolectomy with ileoanal anastomosis (laparoscopy preferred). Sulindac or tamoxifen may help in regression of desmoid tumors.
Gardner’s and Turcot’s syndromes are related to FAP and result from different mutations on APC gene.
Gardner’s syndrome: It presents with colonic polyps, epidermal inclusion cysts, fibromas, lipomas, desmoid tumors and osteomas. It is AD.
Turcot syndrome: It presents with colonic polyps and brain tumors (medulloblastoma with FAP or glioblastoma multiforme with Lynch syndrome). Some cases are associated with Lynch syndrome. It is AD.
MUTYH associated polyposis or MAP: It is an AR polyposis caused by mutations in the MUTYH gene that is involved in DNA repair. Loss of DNA repair leads to acquired mutations in APC gene. Most polyps in MAP are tubular, tubulo-villous or hyperplastic. Most patients develop colon cancer by the age of 70 years. It differs from FAP in that thousands of colorectal polyps develop in adulthood (instead of teenage years). Extraintestinal manifestations are similar to FAP. Screening colonoscopy should be done beginning at age 25-30 years and then every 3-5 years if no polyps are detected. Siblings should also be genetically tested and counselled.
Familial juvenile polyposis: It is an AD disorder that begins in childhood with multiple hamartomatous colorectal polyps. The polyps may progress to form adenomas and colon cancer. It presents with bleeding, intussusception and protein losing enteropathy. Mutations in SMAD4 tumor suppressor gene on chromosome 10q are associated. It may be associated with hereditary hemorrhagic telangiectasia. Congenital abnormalities like malrotation, hydrocephalus, CHD, Meckel’s diverticulum and mesenteric lymphangioma may be present. There is increased risk of colorectal cancers, pancreatic, gastric and duodenal cancers. Colectomy is indicated.
Peutz Jeghers syndrome: It is an AD syndrome characterized by polyposis of the small intestine and less commonly, by colorectal polyps. It is caused by mutations in the STK11, tumor suppressor gene located on chromosome 10p. The polyps are hamartomatous type. Characteristic bluish-black pigmentation of the lips, buccal mucosa, hands, feet and genitalia are seen. The polyps themselves are not pre-malignant but they may cause bleeding, intestinal obstruction or intussusception. There is increased risk for gastrointestinal tumors and cancers of the breast, ovaries, cervix, pancreas, lung, thyroid etc.
Cowden’s syndrome: It is an AD polyposis syndrome caused by mutations in PTEN tumor suppressor gene on chromosome 10q. It presents with hamartomatous polyps in the colon and stomach and extra-intestinal malformations such as thyroid adenomas, goitre, mucocutaneous lesions, fibrocystic disease and fibroadenoma of the breast, uterine fibroids and macrocephaly. There is increased risk for colon, breast and thyroid cancers. Other syndromes associated with PTEN mutations are Bannayan-Riley-Ruvalcaba syndrome and Lhermitte-Duclos syndrome.
Serrated polyposis syndrome: Multiple serrated polyps are present in the colon, which may become malignant. Polypectomy or colon resection is indicated.
Fecal occult blood testing will be positive in polyps and colon cancers. The test has more sensitivity in detecting colon cancer and larger polyps but poor sensitivity to detect small polyps.
Fecal immunochemical testing (FIT) is a more sensitive method to detect occult blood in stool. It detects hemoglobin by using antibodies against globin. FIT is as sensitive as colonoscopy to detect colon cancer but less sensitive than colonoscopy to detect adenomas.
Colonoscopy is the gold standard for diagnosis of colon polyps and cancers. It may miss some lesions, especially smaller ones. Colonoscopy for screening is recommended every 10 years beginning at the age of 50 years. Alternatively, as left colon lesions are more common (especially in Western populations), flexible sigmoidoscopy every 5 years can be used for screening, beginning at age 50 years. If adenomatous or serrated polyps are detected, colonoscopy should be repeated every three years.
Tubular adenoma: low (×400, 2a) and high (×2000, 2b) power magnification views of a hematoxilin-eosin stain. The histological appearance is of branched tubular glands.
Newer techniques such as colonoscopic spectroscopy and narrow-band imaging can be used along with conventional colonoscopy to increase its sensitivity and specificity. CT or MR colonography can be used as non-invasive ways to visualize the colon, but they are inferior to conventional colonoscopy. Capsule endoscopy uses images transmitted by a capsule with tiny cameras that is swallowed by the patient; sensitivity and specificity are less than colonoscopy. Fecal DNA and antigen testing for beta actin, mutant K-ras, BMP3, NDRG4 and hemoglobin are investigational modalities with good sensitivity and specificity.
Polyps >5 mm in size should be resected. Pedunculated polyps can be removed by polypectomy while sessile ones may need a segmental colectomy.
Lynch syndrome or HNPCC (hereditary nonpolyposis colorectal cancer): Most common cause of inherited colon cancer. It is caused by mutations in DNA mismatch repair genes hMLH1, hMSH2 and hMSH6. It is inherited as an AD disorder. Patients have 80% lifetime risk of colorectal cancer. Most colon cancers in HNPCC are located in the right colon. Colon polyps may be seen in Lynch syndrome, contrary to the name H-Non-Polyposis-CC. There is a high risk of tumors in the breast, endometrium, CNS, stomach, small intestine, ovaries and urinary tract.
Screening should begin at the age of 25 years or 5 years earlier than the youngest affected relative and then followed up at one- or two-yearly intervals. Colonoscopy is recommended for screening. Barium enema can be done if colonoscopy is inconclusive. Genetic testing can be done. Immunohistochemistry and screening for microsatellite instability is done on resected tumor samples.
Surgical management includes polypectomy or colectomy, depending on severity/degree of dysplasia. Daily aspirin reduces the risk of colon cancer. Development of other cancers should be monitored by CA-125 levels, endometrial biopsy, neurological examination etc.