Neurodegenerative disorders and dementia
Dementia is the loss of cognitive functioning (thinking, remembering, and reasoning) and behavioral abilities to such an extent that it interferes with a person’s daily life and activities. These functions include memory, language skills, visual perception, problem solving, self-management, and the ability to focus and pay attention. Some people with dementia can’t control their emotions, and their personalities may change.
Delirium is a transient, usually reversible, cause of mental dysfunction. Clinically, it presents with a wide range of neuropsychiatric abnormalities, including waxing and waning levels of consciousness, tremors, illusions, hallucinations, and disorientation.
Stupor occurs in a conscious person who is in a state of psychomotor retardation. They can be aroused only by vigorous physical stimuli such as calling out loudly or prodding.
Coma (unconsciousness) is a state of complete unresponsiveness lasting at least 1 hour. Patients can’t be aroused by vigorous stimulation, although grimacing may occur in response to noxious stimuli. Coma results from gross impairment of both cerebral hemispheres and/or the ascending reticular activating system.
Brain death is the irreversible loss of all functions of the brain, including the brainstem. The three essential findings in brain death are coma, absence of brainstem reflexes, and apnoea. Spinal reflexes may still be present.
(I) Alzheimer’s disease (AD): AD is the most common cause of dementia in older people. It is progressive and interferes with daily activities. Pathological deposits of Abeta amyloid and accumulation of tau protein are seen in AD. Abeta amyloid is derived from APP (amyloid precursor protein) by the action of enzymes called secretases, and it is toxic to neurons. APP is coded by a gene on chromosome 21; therefore, trisomy 21 (Down’s syndrome) carries a high risk of early-onset AD. Mutations in presenilin 1 and 2 genes (PSEN 1 and PSEN 2) on chromosome 14 and 1, respectively, can also cause early-onset AD. The APOE4 allele (chromosome 19) is associated with higher risk for AD, while APOE2 carries decreased risk. Type 2 DM, hypertension, multiple concussions, elevated homocysteine, or chronic inflammation predispose to AD.
Abeta amyloid aggregates as amyloid fibrils, characteristically in the hippocampus and entorhinal cortex of the frontal cortex and medial temporal lobe. Amyloid deposition leads to neuronal cell loss, increased apoptosis, excitotoxicity, synaptic damage, and free radical injury. There is loss of cholinergic neurons in the basal forebrain, decreased acetylcholine levels, and decreased activity of the acetylcholine-synthesizing enzyme choline acetyltransferase in the cerebral cortex.
On gross examination, the brain shows cerebral cortical atrophy, especially in the hippocampal area, while sparing the primary motor, sensory, and visual areas. Gyri appear narrow and sulci widen. The lateral ventricles appear dilated. Amyloid plaques (AP) and neurofibrillary tangles (NT), along with their distribution, support a diagnosis of AD. Histopathological changes first appear in the entorhinal cortex. In severe cases, the brainstem and deep nuclei are involved.
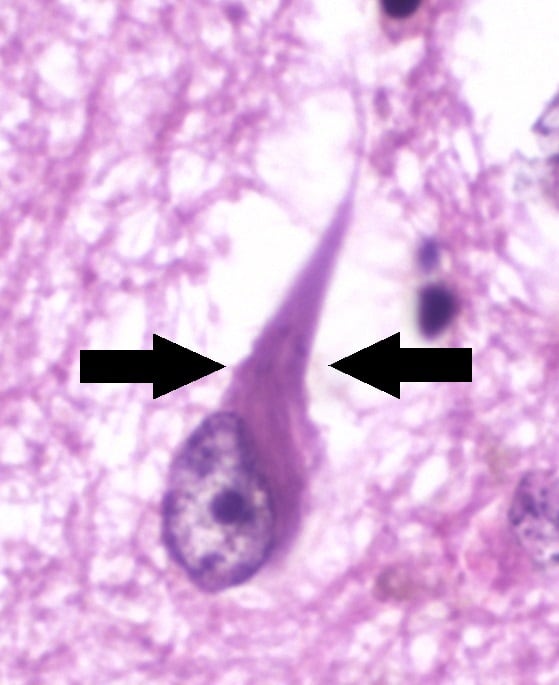
NT are essential for diagnosing AD. They can be seen with special silver stains (modified Bielschowski or Gallyas methods) or by fluorescent or immunohistochemical techniques. NTs appear as parallel, thickened fibrils that surround the nucleus and extend toward the apical dendrite, or as globoid intracytoplasmic structures. They consist of fibrils made of abnormally phosphorylated tau protein. Ubiquitin, cholinesterases, and Abeta amyloid4 are also present. NTs are widespread in layer II neurons of the entorhinal cortex; CA1 and subiculum of the hippocampus; the amygdala; and the deeper layers (layers III, V, and superficial VI) of the neocortex. They correlate directly with disease severity.
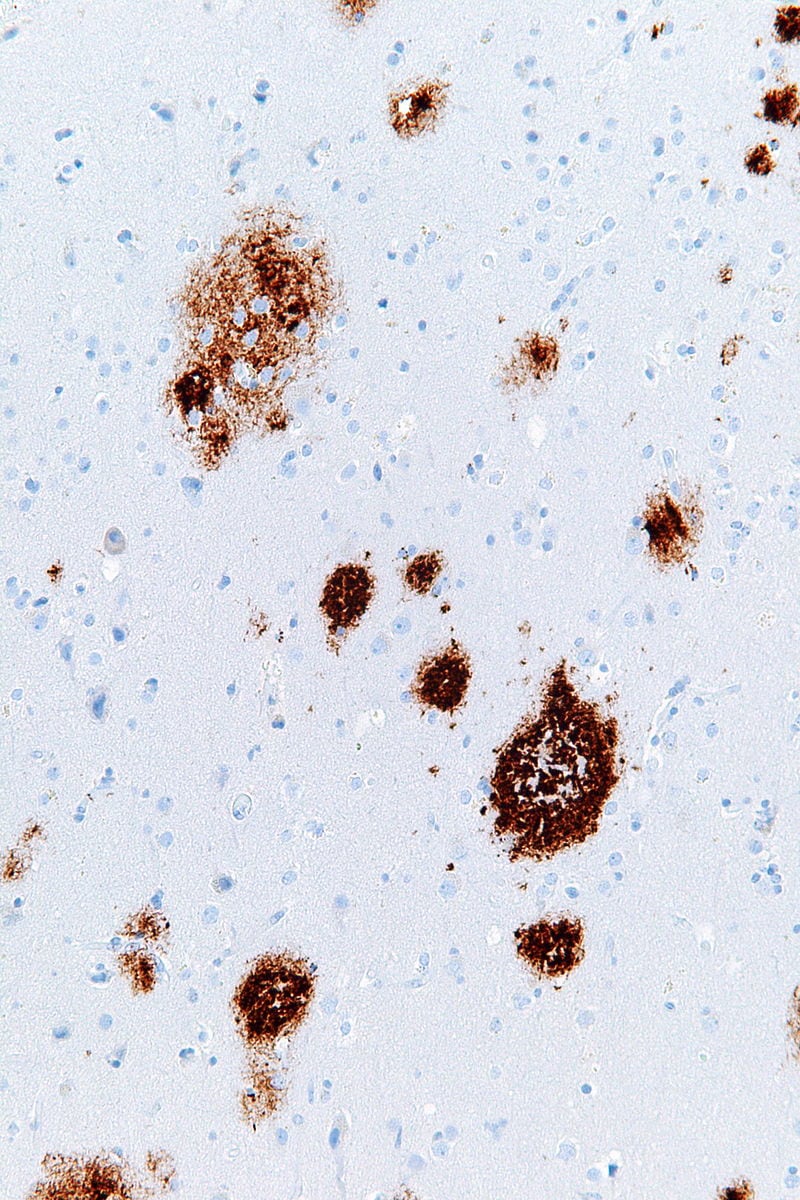
Senile (neuritic) plaques are composed of a central core of beta pleated sheet Abeta amyloid 4 protein arranged radially, surrounded by a corona of neuronal processes. They can be seen with Congo Red staining, silver impregnation techniques, and by polarized microscopy as apple green birefringence. Amyloid angiopathy is also seen and may cause hemorrhages.
Other non-specific lesions include:
- Areas of granulovacuolar degeneration, consisting of granule-containing vacuoles that contain tau protein
- Eosinophilic, rod-like inclusions called Hirano bodies in hippocampal neurons, made of parallel fibres of actin, tropomyosin, and vinculin
Elevation of tau protein and decrease of Aβ in the CSF are useful biomarkers.
(II) Frontotemporal dementia or FTD: Frontotemporal dementia is a group of related conditions resulting from progressive degeneration of the temporal and frontal lobes. It presents as dementia along with personality changes such as apathy, disinhibition, loss of insight and emotional control, language dysfunction, and global cognitive decline. It is the most common cause of dementia in people under the age of 60 years. About 40% of FTLDs are familial, many of them autosomal dominant. Genes involved in autosomal dominant FTDs include MAPT (encodes microtubule-associated protein tau), PGRN (progranulin), and C9ORF72.
Microscopic examination shows neuronal loss and gliosis, vacuolization of the superficial cortex (spongiosis), and ballooned neurons. Abnormal protein deposits are seen not only in neurons but also in glial cells.
| FTD type | Protein inclusion |
| FTD-tau | tau |
| FTD -TDP | TAR DNA binding protein 43 (TDP 43) |
| FTD - FUS | Fused sarcoma protein |
Patients with FTD-TDP develop increasing difficulty understanding the meaning of words and may struggle to find words or name people and objects. People with behavioral variant frontotemporal dementia (bvFTD) often have trouble controlling their behavior. When clinically differentiating FTD from AD, remember that AD typically presents first with dementia, while FTD often presents first with personality changes. In later stages - especially in older patients - it may become impossible to clinically differentiate the two.
(III) Pick’s disease: Pick’s disease is a form of FTD characterized by tau-positive cytoplasmic inclusions called Pick bodies and ballooned neurons with dissolution of chromatin called Pick cells in the fronto-temporal lobes.
(IV) Parkinson’s disease (PD): PD is a common disease of older age. It presents with rigidity, resting tremors (pill rolling), abnormal gait, mask face, slowing of movements, mood disturbances, dementia, and autonomic imbalances. Characteristic gait abnormalities include:
Types of gait in Parkinson’s disease
-
Shuffling gait: Appear to drag their feet while walking
-
Festinating gait: Walking in quick, small steps, shortening of stride
-
Freezing of gait: Temporary problems in starting to walk, feels like feet are glued to the floor.
Some cases of PD are inherited in AD or AR fashion. Mutations of α-synuclein, a synaptic protein encoded by SNCA on chromosome 4q22, are linked to the AD form of PD. Mutations in the Parkin gene have been associated with AR PD. A few patients have GBA1 mutations associated with Gaucher disease; patients with Gaucher disease are prone to develop PD, with earlier onset and more severe involvement. Most cases are idiopathic. Environmental neurotoxins such as the meperidine derivative MPTP, phenothiazines, arsenic, chronic CO poisoning, and Wilson’s disease can cause PD.
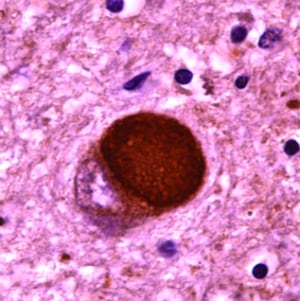
The primary pathology is deposition of alpha synuclein, which damages dopamine-secreting neurons in the substantia nigra. Under physiological conditions, the main function of α-synuclein is control of neurotransmitter release through effects on the SNARE complex. In PD, alpha synuclein forms abnormal protofibrils that become insoluble polymers and disrupt cellular homeostasis. Its accumulation impairs mitochondrial, lysosomal, and endoplasmic reticulum function, and it interferes with microtubule transport and synaptic transmission. Alpha synuclein deposits as round, lamellated eosinophilic cytoplasmic inclusions called Lewy bodies and as fibrils called Lewy neurites in astrocytes and oligodendroglial cells. Lewy bodies are found not only in the substantia nigra but also in other parts of the CNS and PNS, including the cerebral cortex, reticular activating system, limbic system, olfactory pathways, and nucleus basalis of Meynert. On gross examination, there is loss of pigmentation of the substantia nigra. Depigmentation occurs because melanin synthesis is affected due to death of dopamine-producing neurons. Decreased dopamine contributes to rigidity and movement problems in PD.
Lewy body disease or dementia: Lewy body disease is pathophysiologically and clinically similar to Parkinson’s disease. They are differentiated by the timing of cognitive and movement symptoms. In dementia with Lewy bodies, cognitive symptoms develop within a year of parkinsonism, while in Parkinson’s disease dementia, cognitive symptoms develop more than a year after the onset of movement symptoms. Visual hallucinations and REM sleep behavior disorder (acting out dreams in sleep) are seen in Lewy body dementia.
(V) Progressive supranuclear palsy or Steele-Richardson-Olszewski syndrome: Progressive supranuclear palsy is a condition that causes changes in movement, language, and behavior. In its typical form, PSP causes difficulties with balance that lead to frequent falls. Eye movements are impaired. Involuntary blepharospasm and eye opening apraxia (inability to open an eye) are seen. Other patients have trouble closing their eyes or may blink less than normal, causing the eyes to become dry and red. Abnormal deposition of tau protein is seen in neurons. Some patients have mutations in the MAPT gene that codes for tau protein.
(VI) Huntington’s disease (HD): HD is an autosomal dominant inherited condition presenting with involuntary jerking or twitching movements (chorea), abnormal body postures, and changes in behavior, emotion, judgment, cognition, or dementia. People with HD also develop impaired coordination, slurred speech, and difficulty feeding and swallowing. HD typically begins between ages 30 and 50. An earlier-onset form called juvenile HD occurs under age 20. Its symptoms differ somewhat from adult-onset HD and include rigidity, slowness, difficulty at school, rapid involuntary muscle jerks called myoclonus, and seizures. HD is caused by a mutation in the HTT gene that codes for a protein called huntingtin. The defect involves cytosine, adenine, and guanine repeats (CAG repeats). Normally, the CAG segment is repeated 10 to 35 times within the gene. In people with Huntington disease, the CAG segment is repeated 36 to more than 120 times. People with 36 to 39 CAG repeats may or may not develop signs and symptoms of HD, while people with 40 or more repeats almost always develop the disorder. Huntingtin deposits in neurons precipitate cell death.
Gross examination of the brain reveals atrophy of the caudate nucleus and putamen and dilatation of the anterior horns of the lateral ventricles. Cortical atrophy also occurs, but the medial temporal lobes are spared. Microscopic examination shows loss of medium-sized spiny, enkephalin-containing internuncial neurons in the caudate nucleus and putamen, loss of cortical neurons, and gliosis. Reactive proliferation of astrocytes is seen. Intranuclear inclusions containing huntingtin are present in neurons. The striatum shows loss of GABA-, acetylcholine-, and glutamate-producing neurons. Decreased GABA and unbalanced dopamine activity result in chorea.
(VII) CJD: Creutzfeldt-Jakob disease (CJD) is a rapidly progressive, invariably fatal neurodegenerative disorder believed to be caused by an abnormal isoform of a cellular glycoprotein known as the prion protein. It is a subtype of human prion diseases.
Types of human Prion diseases
| Clinical features | CSF 14-3-3 protein | Histopath findings | EEG and MRI findings | |
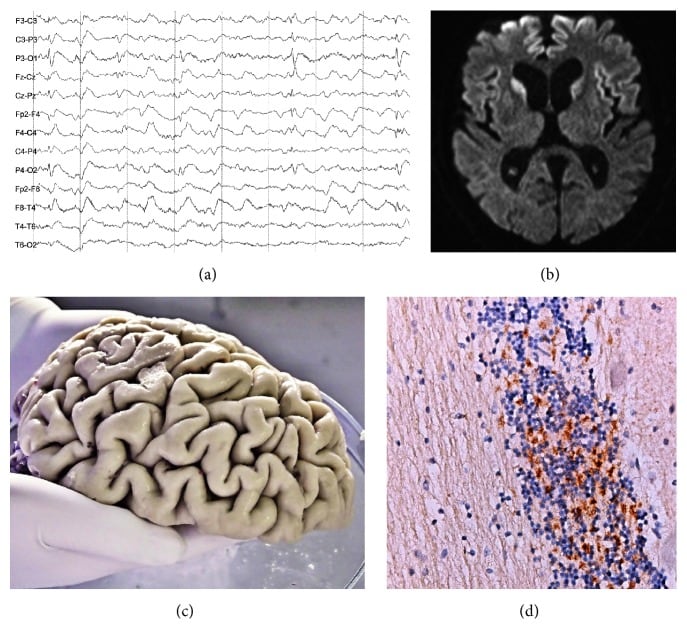
| Sporadic CJD | Progressive dementia, myo-clonus, cerebellar ataxia, visual problems, extrapyramidal symptoms | Positive | Spongiform changes, neuronal loss, astrogliosis, PrP- deposition | Brain atrophy and hyperintensities in basal ganglia and cortex on MRI. EEG shows periodic sharp wave complexes. |
| Inherited CJD | Similar to sporadic CJD. | Positive | Same as above | Same as above |
| Variant CJD | Onset in younger age group, depression, anxiety, social withdrawal, dementia. | Positive | Same as above plus PrP- deposits as florid plaques | Hyperintensities in the posterior thalamus, Pulvinar sign positive (thalamus); Non-specific EEG changes |
| GSS or Gerstmann-Straussler-Scheinker syndrome | Cerebellar dysfunction presenting as ataxia, nystagmus, dysarthria. | Negative | Same as above except for PrP- deposition in the form of multicentric plaques | Normal or cerebral or cerebellar atrophy; Non-specific EEG changes |
| FFI or fatal familial insomnia | Insomnia, autonomic dysfunction | Negative | Involvement of the thalamus | Normal or cerebral or cerebellar atrophy; Non-specific EEG changes |
| Iatrogenic | Similar to sporadic CJD | Positive | Same as sCJD | Same as sCJD |
A familial form of CJD, Gerstmann-Sträussler-Scheinker syndrome, and fatal familial insomnia are inherited human prion diseases. They are inherited in an autosomal dominant manner and are associated with mutations in the PRNP gene, which codes for the prion protein. Iatrogenic CJD can occur after implantation of human dura mater, corneal graft implantation, or treatment with human cadaveric pituitary extracts. Variant CJD represents transmission of BSE (bovine spongiform encephalopathy, or mad cow disease) prions to humans.
All prion diseases involve abnormal folding of prion protein. Normal prion protein (PrPc) is converted to abnormal PrPSc, which is resistant to proteolytic proteases and deposits in cells as an insoluble amyloid. Prions are self-propagating and transmissible: once PrPSc is generated endogenously or introduced from the environment, it converts normal prion proteins into abnormal ones. Affected cells ultimately die. There is no associated immune or inflammatory response.