Neoplasms
Primary CNS tumors are more common in children, while metastases are a more common cause of brain tumors in adults. Supratentorial (cerebral) tumors are more common in adults, while infratentorial tumors (cerebellum, brainstem) are more common in children.
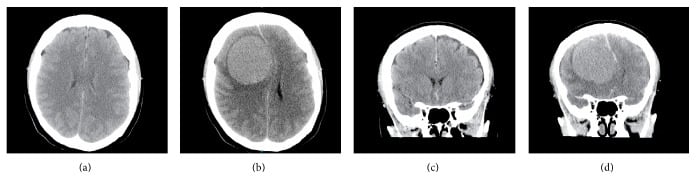
(I) Meningiomas: These are benign, slow-growing tumors that arise from the arachnoid mater. They’re most commonly seen in adult women, and they grow faster during pregnancy. Common locations include the lateral cerebral convexities, the midline along the falx cerebri, and the olfactory groove. They may also occur in the cerebral ventricles or the spinal cord. There is an increased risk of multiple meningiomas in neurofibromatosis type 2.
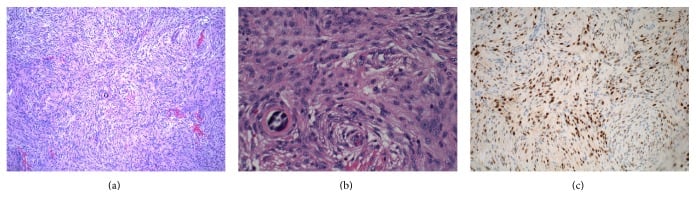
On histopathological examination, meningiomas are well-circumscribed, solid, spherical masses attached to the dura. On cut section, they are firm and fibrotic, often with foci of calcification. Characteristic Psammoma bodies are seen microscopically.
Various histologic patterns may be seen:
- Syncytial: masses of polygonal cells with poorly defined cell membranes
- Fibrous: spindle-shaped cells arranged in parallel or interlacing bundles
- Transitional (mixed): a mix of the previous two patterns, with conspicuous Psammoma bodies
- Hemangioblastoma-like pattern: associated with a high rate of recurrence
- Anaplastic pattern: a rare malignant form that invades surrounding structures and metastasizes to the lung
Most meningiomas show deletion of chromosome 22.
(II) Neurofibromas: These are benign tumors arising from Schwann cells in the PNS. They may be single or multiple. Multiple cutaneous neurofibromas are seen in neurofibromatosis type 1 (Von Recklinghausen disease).
Clinical features of neurofibromatosis types 1 and 2
NF type 1
- Multiple cutaneous neurofibromas
- Plexiform tumors (may become malignant)
- Optic gliomas
- Lisch nodules (hamartomas of the iris)
- Cafe au lait (coffee brown color) macules
- Axillary and inguinal freckling
- Sphenoid dysplasia and fibromuscular dysplasia of the arteries
NF type 2
- Bilateral vestibular Schwannomas
- III or V nerve Schwannomas
- Hearing loss, dizziness, headaches, diplopia, facial weakness
- Meningiomas, CNS gliomas
- Spinal ependymomas
- Cutaneous neurofibromas
Neurofibromatosis is inherited as an autosomal dominant syndrome.
- Type 1 involves mutations in the NF 1 gene on chromosome 17. NF 1 is a tumor suppressor gene that normally codes for neurofibromin, an inhibitor of the ras/MAPK pathway.
- Type 2 involves mutations in the NF 2 gene on chromosome 22. NF 2 is a tumor suppressor gene that normally codes for merlin. Merlin regulates cell growth in Schwann cells by inhibiting MAPK signalling.
On histopathology, neurofibromas are un-encapsulated tumors that produce a fusiform enlargement of the nerve. They show bundles of interlacing fascicles of spindle-shaped cells with wavy nuclei in a collagenous and mucoid matrix. They stain positive for S-100 and EMA (epithelial membrane antigen) by immunohistochemistry.
(III) Astrocytoma and glioblastoma multiforme: Astrocytoma is a type of glioma; other gliomas include ependymoma, glioblastoma, and oligodendroglioma. Gliomas arise from neuroglial tissue. Astrocytoma and glioblastoma multiforme arise from astrocytes.
WHO grading system for astrocytomas
| WHO Grade | Histopathological features |
| Grade I-Pilocytic astrocytoma | Benign cytological features - see below |
| Grade II-Diffuse astrocytoma | Moderate cellularity - no anaplasia or mitotic activity |
| Grade III- Anaplastic astrocytoma | Cellularity, anaplasia, mitoses |
| Grade IV-Glioblastoma multiforme | Same as Grade III plus microvascular proliferation and necrosis |
Grade I is the least malignant, while Grade IV is the most malignant.
- Grade I (pilocytic) is low grade and has a good prognosis. It’s more common in children and young adults and shows wavy, fibrillary processes; some cases have pleomorphic features.
- Grade II is also fibrillary, composed of well-differentiated astrocytes, and is commonly seen in middle age.
- Grade III is anaplastic and hypercellular, with nuclear atypia, hyperchromasia, and characteristic vascular proliferation.
- Grade IV (glioblastoma multiforme) is very aggressive. Grossly, it appears as grey-white tissue with yellow necrotic areas and interspersed hemorrhages. Histology shows palisaded layers of tumor cells, anaplasia, fusiform cells, and small round poorly pleomorphic cells. Hypoxia-driven vascular proliferation with glomerulus-like structures is seen. Glioblastoma is most common in middle-aged adults, typically in the frontal and temporal lobes.
All astrocytomas stain positive for GFAP (glial fibrillary acidic protein) on immunohistochemistry. Imaging in glioblastoma shows a large irregular mass of variable density with cavitation, surrounded by a large area of edema. It may spread across the corpus callosum from one hemisphere to the other.
(IV) Medulloblastoma: This is a highly malignant tumor of embryonal origin. It’s seen most commonly in small children and sometimes in young adults. It is located in the cerebellum or around the fourth ventricle. Patients with Turcot’s syndrome are at increased risk of developing medulloblastomas.
It invades locally and can show distant metastases to the lungs, liver, vertebrae, and pelvis. It can obstruct the fourth ventricle, causing hydrocephalus. Microscopically, it shows typical Homer-Wright rosettes, consisting of small, poorly differentiated cells arranged around blood vessels. Cells may show glial, neuronal, and other differentiation (such as striated muscle and melanocytes). Some tumors may show extensive collagen production.
(V) Ependymoma: This tumor is derived from the ependymal lining of the ventricles or the central canal. It is more common in children and young adults, most commonly in the fourth ventricle. Microscopically, ependymal cells can form canaliculi, rosettes, and pseudorosettes. They may present with hydrocephalus.
(VI) Primary CNS lymphoma: This is most commonly seen in immunocompromised adults above age 50, such as organ transplant recipients or AIDS patients. They arise from microglia/histiocytes or lymphocytes. They are usually periventricular, frequently involve the corpus callosum, and can spread to both cerebral hemispheres. Tumor cells form dense perivascular sheaths or diffuse masses and are histologically similar to diffuse large cell lymphomas.
(VII) Craniopharyngioma: This is a benign, suprasellar tumor derived from epithelial remnants of Rathke’s pouch. It is more common in children and adolescents and is located in the region of the optic chiasma. It forms a reddish, cystic mass composed of sheets of squamous epithelial cells and keratin, set in a loose connective tissue stroma with areas of calcification. Cholesterol crystals are seen.
Craniopharyngiomas present clinically with headaches, bitemporal hemianopsia, decreased visual acuity, papilledema, and endocrine dysfunction due to compression of the hypothalamus and pituitary stalk. GH, GnRH, TSH, ACTH, and ADH deficiencies may be seen.
(VIII) Metastatic tumors: These are more common than primary tumors. The most common tumors that metastasize to the brain include lung, breast, melanomas, colon, and renal. ALL (acute lymphoblastic leukemia) frequently involves the brain. Breast cancer metastases can develop late, sometimes years after the diagnosis of the primary cancer.
Metastatic tumors to the brain are often:
- Multiple
- Supratentorial
- Spread by the hematogenous route
- Well demarcated
- Located at the grey-white matter interface
Microscopically, they resemble the primary tumor. They typically present with symptoms of cerebral edema, increased intracranial pressure, seizures, and focal deficits.
Meningeal carcinomatosis is a condition in which secondary metastases diffusely involve the brain and spinal cord, presenting as multiple nodular growths. The syndrome results from tumor infiltration and inflammation of the leptomeninges. Access to the subarachnoid space occurs either by hematogenous dissemination or by direct extension from a tumor of the brain or spinal cord. It is seen in carcinoma of the lung, breast, and ALL. It presents with headache, drowsiness, cranial nerve deficits, spinal root pain, and paresthesias. The CSF in meningeal carcinomatosis shows high protein, low glucose, neoplastic cells, and lymphocytes.