Pharmacokinetic drug-drug interactions
Pharmacokinetics is “what the body does to the drug.” It includes absorption, distribution, metabolism, excretion, and overall drug elimination.
A helpful way to organize pharmacokinetic interactions is the ADME framework:
- “A” for absorption
- “D” for distribution
- “M” for metabolism
- “E” for elimination
Drug absorption
Drug absorption depends on the route of administration and the drug’s properties.
In general, small, unionized, lipid-soluble drugs are absorbed more easily. Most drugs are absorbed in the small intestine because it has a large surface area.
Most drugs are absorbed by passive diffusion, meaning they move from an area of higher concentration to an area of lower concentration. Some drugs use other mechanisms, including active transport, facilitated passive diffusion, and pinocytosis.
Ionization matters because unionized drug molecules cross membranes more easily:
- Weak acids are more unionized in an acidic environment, so they’re absorbed better in acidic conditions.
- Weak bases are more unionized in an alkaline environment, so they’re absorbed better in alkaline conditions.
This same idea helps explain urinary drug excretion: alkalinizing the urine can accelerate the excretion of weak acids (like aspirin), and acidifying the urine can accelerate the excretion of weak bases.
Drug distribution
Drug distribution is not uniform throughout the body. Several factors determine how much drug reaches different tissues:
- Solubility: Lipid-soluble drugs (for example, some anesthetics) can cross into lipid-rich tissues such as the brain.
- Protein and tissue binding: Binding affects where the drug “stays.” For example, warfarin largely remains in the bloodstream because it binds strongly to plasma albumin.
- Blood supply (perfusion): Well-perfused tissues reach higher drug concentrations faster than tissues with lower blood flow.
- Tissue mass: The amount of tissue can influence how much drug is taken up overall.
- Type of protein binding:
- Acidic drugs tend to bind more to albumin.
- Basic drugs tend to bind more to alpha-1 acid glycoprotein and lipoproteins.
Protein binding influences both distribution and duration of action because only the unbound fraction can readily leave the bloodstream and interact with targets.
Volume of distribution (Vd) describes how extensively a drug distributes in the body:
- Drugs with a small Vd are largely confined to the blood.
- Drugs with a large Vd distribute widely into tissues and body compartments.
Drug metabolism
Drug metabolism occurs primarily in the liver, but it can also occur in the kidneys, lungs, blood, and intestines.
Key points about metabolism:
- Prodrugs are metabolized into active drugs.
- Most drugs become inactive after metabolism.
- Some drugs form toxic metabolites.
- A major goal of metabolism is to make the drug (or its products) more water soluble (polar) so it can be eliminated more easily.
Types of metabolic reactions:
- Phase 1 reactions (non-synthetic): Convert the drug to a more polar or more reactive/active form. Oxidation, reduction, deamination, hydrolysis.
- Phase 2 reactions (synthetic): Make the drug more water soluble; the end product is inactive. Conjugation with glucuronic acid, glycine, sulfate, glutathione, etc.
Metabolism rates vary among patients:
- Rapid metabolizers may not reach effective blood and tissue levels.
- Slow metabolizers have a higher risk of adverse effects.
Factors that affect metabolism include genetics, age, coexisting liver or other diseases, and concomitant use of drugs that alter metabolism.
Cytochrome P450 (Cyt P450) enzymes (primarily on the smooth endoplasmic reticulum of the liver) carry out most oxidation reactions. Humans have approximately 60 cytochrome P450 genes, and polymorphisms can change drug metabolism. Major enzymes include CYP3A4 and CYP2D6.
- Inhibitors of Cyt P450 decrease enzyme activity.
- Inducers of Cyt P450 increase enzyme activity.
Common inhibitors and inducers of Cyt P450 enzymes:
-
Inhibitors: Cimetidine, amiodarone, erythromycin, ticlopidine, ciprofloxacin, omeprazole, fluconazole, bupropion, metoclopramide, quinidine, disulfiram, metronidazole, grapefruit juice, ritonavir, saquinavir. Need to lower the dose of a drug given concomitantly with any drug (s) in this group.
-
Inducers: Carbamazepine, phenytoin, rifampin, phenobarbital, isoniazid, alcohol, tobacco, dexamethasone, prednisone. Need to increase the dose of a drug given concomitantly with any drug (s) in this group.
Drug excretion or elimination
Most drugs are excreted by the kidneys. Inhaled anesthetics are mainly excreted by the lungs, and some drugs are excreted in bile and feces.
Elimination ends drug activity by converting the drug to an inactive metabolite. Drugs undergo two types of elimination: first-order and second-order.
First-order elimination occurs when the rate of elimination is directly proportional to the remaining drug concentration. In other words, a constant fraction of the drug is eliminated per unit time.
- These drugs have a constant half-life.
- A plasma concentration vs. time graph shows an exponential decrease.
- Drug concentration decreases by 50% with each half-life.
- Most drugs follow first-order kinetics.
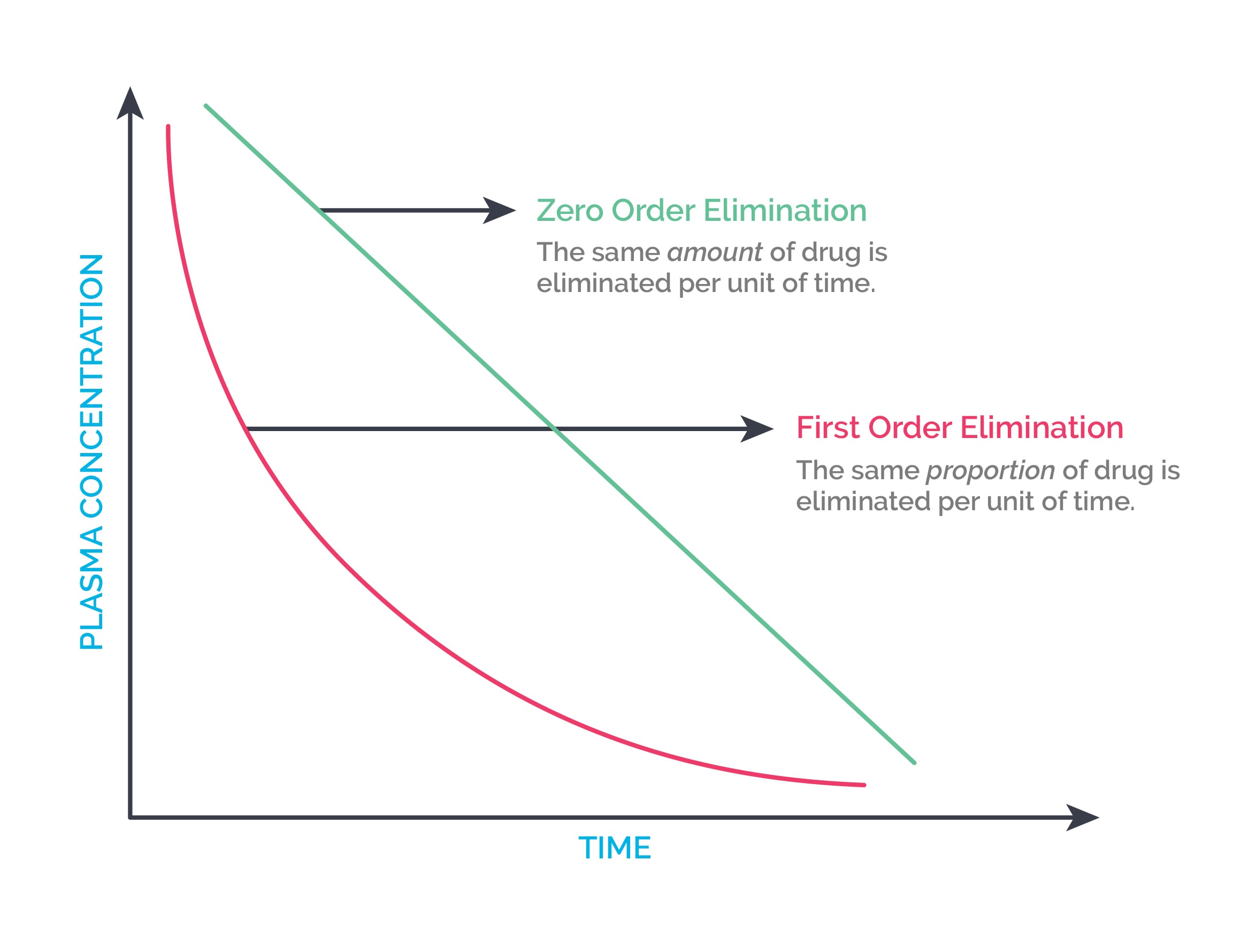
Zero-order elimination occurs when a constant quantity of drug is eliminated per unit time.
- This happens when elimination mechanisms become saturated.
- A plasma concentration vs. time graph shows a linear decrease.
- There is no constant half-life.
Ethanol, aspirin, phenytoin at high doses, omeprazole, fluoxetine, and cisplatin show zero-order elimination.
| Example | ADME level | Mechanism |
|---|---|---|
| (Tetracyclines or quinolones) + multivalent cations (calcium, magnesium, or aluminum) | Absorption | Reduces absorption by the formation of complexes that cannot be absorbed |
| Itraconazole + antacids | Absorption | Increased pH minimizes the absorption of acidic drugs |
| Cyclosporine + St. John’s wort | Absorption | St. John’s wort increases the activity of P-glycoprotein* in the intestinal cells and decreases cyclosporine levels |
| Phenytoin + valproic acid | Distribution | Both drugs compete for binding sites on albumin |
| Rifampin + oral contraceptive pills | Metabolism | Rifampin induces Cyt P450, causing decreased estrogen levels |
| Warfarin + grapefruit juice | Metabolism | Grapefruit juice inhibits Cyt P450, causing increased warfarin levels, which may cause bleeding |
| Penicillin + probenecid | Elimination | Increased penicillin levels as probenecid competes with penicillin for transporters in the kidney |
*P-glycoprotein is a membrane transporter that transports substances out of the cell. Certain drugs, supplements, and foods activate or inhibit P-glycoprotein, which can decrease or increase the levels of medications taken along with them.