Pharmacokinetics
Pharmacokinetics: Pharmacokinetics is the study of “what the body does to the drug.” It includes absorption, distribution, metabolism, excretion, and elimination of the drug.
Drug absorption: Drug absorption depends on the route of administration and the drug’s own properties. In general, small, unionized, lipid-soluble drugs are absorbed more easily.
Weak acids are more unionized in an acidic environment, so they’re absorbed better in acidic conditions. Weak bases are more unionized in an alkaline environment, so they’re absorbed better in alkaline conditions. For the same reason, excretion of weak acids (like aspirin) can be accelerated by alkalinizing the urine, and vice versa.
Most drugs are absorbed in the small intestine because it has a large surface area. Most drugs are absorbed by passive diffusion (movement from an area of high concentration to an area of low concentration). Other mechanisms include active transport, facilitated passive diffusion, and pinocytosis.
Bioavailability is the proportion of a dose that reaches the systemic circulation. The first-pass effect is metabolism of a drug into inactive forms, which decreases bioavailability. It occurs in the liver, intestines, etc. Orally administered drugs must pass through the intestinal wall and then the portal circulation to the liver, so bioavailability of orally administered drugs is often low.
Bioavailability is affected by route of administration, age, sex, previous GI surgery, effect of food, physicochemical properties, gastric emptying rate, intestinal transit time, blood flow, GI flora, and enzymes.
Less absorption = low bioavailability High first-pass metabolism = low bioavailability
Drug distribution: Drug distribution is uneven in the body, meaning some tissues reach higher drug concentrations than others. Distribution is affected by:
- Solubility of the drug (e.g., lipid-soluble drugs like some anesthetics can easily enter the lipid-rich brain)
- Tissue binding (e.g., warfarin stays mostly in the bloodstream because it binds to plasma albumin)
- Blood supply (drug concentration rises more rapidly in richly perfused tissues than in poorly vascular tissues)
- Total mass of the tissue
Acidic drugs are usually bound more extensively to albumin. Basic drugs are usually bound more extensively to alpha-1 acid glycoprotein, lipoproteins, or both. Tissue binding prolongs the duration of action of a drug.
Volume of distribution (Vd) is a measure of how a drug is distributed in the body.
Vd = Dose/plasma concentration
Drugs with a small Vd are largely confined to the blood. Drugs with a large Vd are extensively distributed throughout body compartments and tissues. Drugs with a large Vd generally need higher doses to achieve a target plasma concentration, and vice versa. Similarly, lipophilic drugs tend to have a larger Vd.
Drug metabolism: Drug metabolism occurs primarily in the liver. Other sites include the kidney, lung, blood, and intestines. Prodrugs are metabolized to form active drugs. Most drugs become inactive after metabolism, although some drugs produce toxic products during metabolism.
A key goal of metabolic reactions is to make the drug (or its products) more water soluble (more polar) so it can be eliminated more easily.
Types of metabolic reactions
| Phase 1 reactions (non-synthetic) | Phase 2 reactions (synthetic) |
| Converts drug to more polar or more reactive/active form | Make drug more water soluble, end product is inactive |
| Oxidation, reduction, deamination, hydrolysis | Conjugation with glucuronic acid, glycine, sulfate, glutathione etc. |
Drug metabolism rates vary among patients. In rapid metabolizers, effective blood and tissue levels of the drug may not be reached. In slow metabolizers, the risk of adverse effects is higher. Metabolism rate is affected by genetics, age, coexisting liver or other diseases, and concomitant use of drugs that affect metabolism.
Most oxidation reactions are carried out by cytochrome P450 enzymes, located primarily on the smooth endoplasmic reticulum of the liver. There are approximately 60 cytochrome P450 genes in humans. NADPH acts as an electron donor to cytochrome P450 enzymes. Polymorphisms in cytochrome P450 enzymes affect drug metabolism. Major ones are CYP3A4 and CYP2D6.
Common inhibitors and inducers of cyt P450 enzymes
| Inhibitors | Inducers |
| Cimetidine, amiodarone, erythromycin, ticlopidine, ciprofloxacin, omeprazole, fluconazole, bupropion, metoclopramide, quinidine, disulfiram, metronidazole, grapefruit juice, ritonavir, saquinavir | Carbamazepine, phenytoin, rifampin, phenobarbital, isoniazid, alcohol, tobacco, dexamethasone, prednisone, |
| Need to lower the dose of a drug given concomitantly with any drug (s) in this group | Need to increase the dose of a drug given concomitantly with any drug (s) in this group |
Glucuronidation is the most common conjugation reaction. The main site is the liver microsomal system. In glucuronidation, the enzyme UDP-glucuronosyltransferase conjugates the drug with glucuronic acid, converting it into hydrophilic and negatively charged glucuronides that can be excreted easily.
This process is slower in newborns, which puts them at risk of drug toxicity (e.g., from chloramphenicol). Drugs like acetaminophen, diazepam, digoxin, morphine, and sulfamethoxazole undergo glucuronidation.
Drug excretion: Most drugs are excreted by the kidney. Inhaled anesthetics are mainly excreted by the lungs. Some drugs are excreted in bile and feces.
Elimination is the conversion of a drug to an inactive metabolite. Elimination leads to termination of drug activity. Drugs undergo two types of elimination: first order and second order.
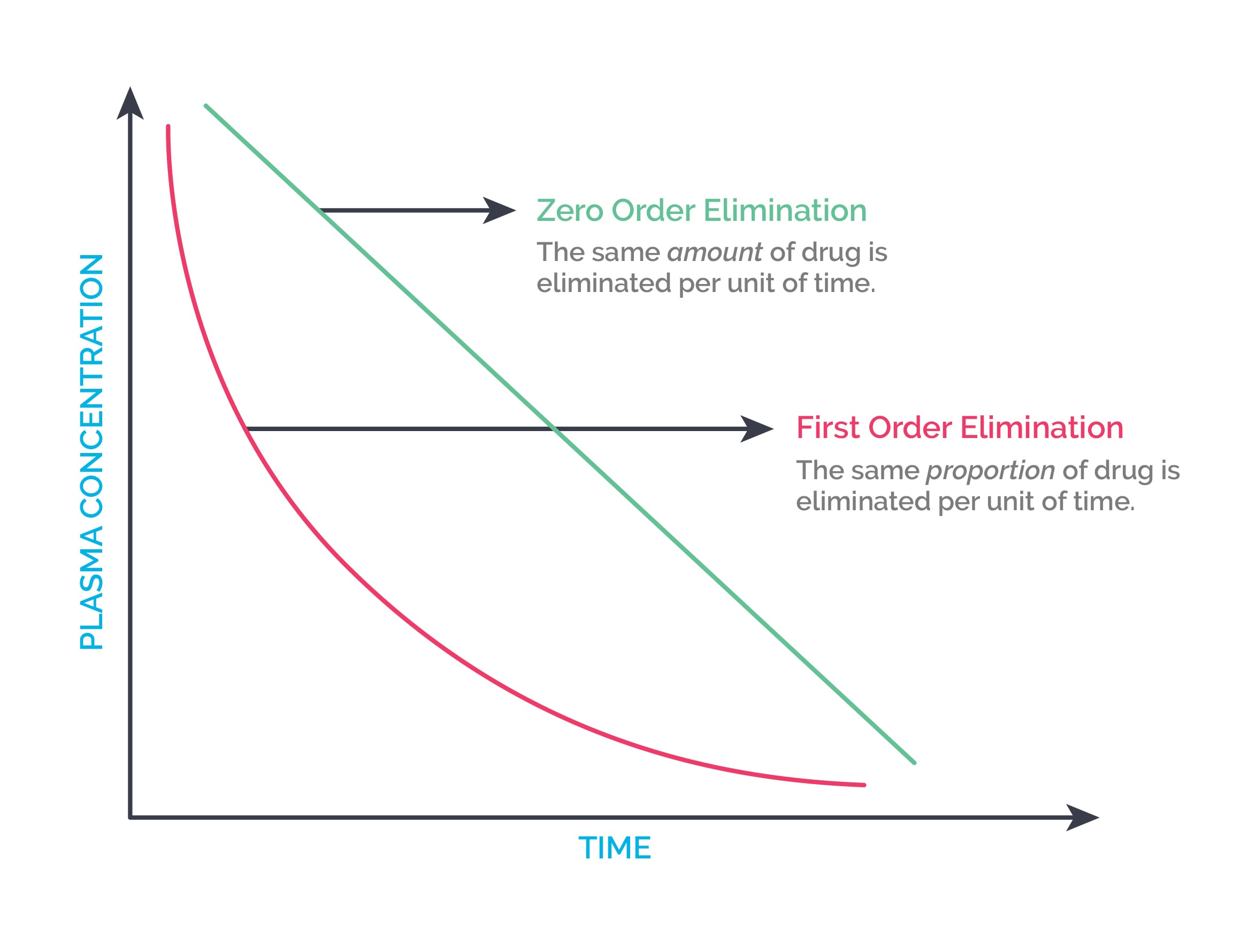
First-order elimination means the rate of elimination is directly proportional to the remaining concentration of the drug. In other words, a constant fraction of the drug is eliminated per unit time. These drugs have a constant elimination half-life. They show an exponential decrease in plasma concentration when plasma concentration is plotted against time. Drug concentration decreases by 50% each half-life. Most drugs follow first-order kinetics.
Zero-order elimination means a constant quantity of the drug is eliminated per unit time. Drugs that saturate their elimination mechanisms show zero-order kinetics. They show a linear decrease in plasma concentration when plasma concentration is plotted against time. They do not have a constant half-life. Ethanol, aspirin, phenytoin at high doses, omeprazole, fluoxetine, and cisplatin show zero-order elimination.
Clearance of drug: It is given by the formula below.
Clearance = Rate of elimination of the drug / Concentration of the drug in plasma
Clearance is constant for drugs that follow first-order kinetics, but it is not constant for drugs that follow zero-order kinetics.
Half life of drug (t1/2): Half-life is the time it takes for the concentration of a drug in the body to decrease by half. For example, if 5 hours are required to decrease the concentration of drug A from 100 mg/dl to 50 mg/dl, then the half-life is 5 hours. It is given by the formula below.
t ½ = 0.693 X Vd/ Clearance
Vd is the volume of distribution.
Any factor that affects Vd or clearance will affect half-life. Age and renal failure (check creatinine clearance or GFR) are the most common factors that affect the half-life of a drug.
3 to 4 half-lives are needed for a drug given by continuous infusion to reach steady-state concentration (i.e., stable or constant plasma drug concentration).
Loading dose: It is calculated by the following formula.
Ld = Vd X desired plasma concentration / bioavailability
Loading dose is not affected by clearance.
Maintenance dose: It is given by the following formula.
Md = Clearance X desired plasma concentration/ bioavailability
If the volume of distribution changes, the drug dose should be changed. If the elimination half-life of the drug changes, the dosing interval should be changed.
Therapeutic window: It is the safe range between the minimum therapeutic concentration and the minimum toxic concentration.
Therapeutic index (TI) or therapeutic ratio: It is the ratio of the dose that produces toxicity to the dose needed to produce the desired therapeutic response. It is given by the following formula. Drugs with a narrow TI need to be meticulously monitored because the difference between their therapeutic and toxic doses is very small.
TI = TD50/ED50, where TD50 is the toxic dose and ED50 is the effective dose for 50% of population tested. Higher the TI, safer is the drug.
Drugs with a narrow therapeutic index
- Digoxin
- Lithium
- Phenytoin
- Theophylline
- Warfarin