Complement deficiencies
Hereditary angioedema (C1 esterase inhibitor deficiency): This is an AD disorder characterized by recurrent episodes of severe swelling (angioedema) affecting the limbs, face, intestinal tract, and airway. Episodes may be precipitated by trauma or stress, and sometimes occur without a clear trigger. Typical features include severe abdominal pain, nausea, vomiting, airway obstruction, and erythema marginatum. Mutations in the SERPING1 and F12 genes are seen. Decreased levels or abnormal activity of C1 inhibitor leads to increased bradykinin levels, which promotes inflammation and edema.
PNH (paroxysmal nocturnal hemoglobinuria): This is an acquired disorder characterized by paroxysmal episodes of hemolysis and hemoglobinuria. Episodes may be triggered by physiologic stress, such as infections or physical exertion. Relatively lower oxygen levels during sleep predispose to hemolysis. Hemoglobinuria presents as passing brown or cola-colored urine, which is most noticeable in the morning when urine has accumulated overnight (nocturnal). Other symptoms include fatigue, weakness, pallor, shortness of breath, tachycardia, recurrent infections due to leukopenia, recurrent thrombosis (or rarely hemorrhage), and an increased risk of leukemia.
Mutations in the PIGA gene are seen in hematopoietic stem cells. The PIGA gene codes for phosphatidylinositol glycan class A, which is required for the synthesis of GPI, a cell membrane anchor. DAF (decay accelerating factor) or CD55 and CD59 are anchored on the RBC surface with the help of GPI. CD55 and CD59 protect RBCs from complement-mediated lysis. In PNH, deficiency of these molecules leads to increased complement activation and hemolysis. RBCs, WBCs, and platelets are mainly affected.
Deficiency of phagocytic cells
Chediak-Higashi syndrome: This is an AR disorder characterized by recurrent pyogenic infections, oculocutaneous albinism, nystagmus, photophobia, bleeding tendencies, seizures, ataxia, and peripheral neuropathy. Chediak-Higashi syndrome is caused by mutations in the LYST gene, which provides instructions for making the lysosomal trafficking regulator. This protein plays a role in trafficking materials into lysosomes. In this condition, neutrophil lysosomes fail to fuse with phagosomes. Superoxide levels are normal. Large granular inclusions are seen in cells. Melanin is trapped within large melanosomes, causing albinism.
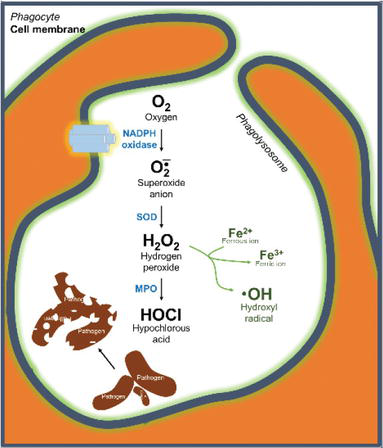
Chronic granulomatous disease (CGD): This is an X-linked (sometimes AD) disorder characterized by increased infections with catalase-positive bacteria (e.g., S. aureus), gram-negative enteric bacteria (e.g., Serratia and Burkholderia), and Aspergillus fumigatus. It is caused by defective intracellular microbicidal activity of neutrophils due to a lack of NADPH oxidase. As a result, no H2O2 or superoxides are formed (no oxidative burst). Granulomas can form in tissues and may obstruct the GI tract or bladder. Pneumonia, fungal infections, and skin, liver, and lymph node lesions are seen. Diagnosis is made by the NBT (nitroblue tetrazolium) test, which will be negative due to absence of the respiratory burst (phagocytic granules do not turn deep blue), or by a dichlorofluorescein assay using flow cytometry. Cure is an allogeneic hematopoietic stem cell transplantation.
Leukocyte adhesion deficiency is discussed in another chapter.