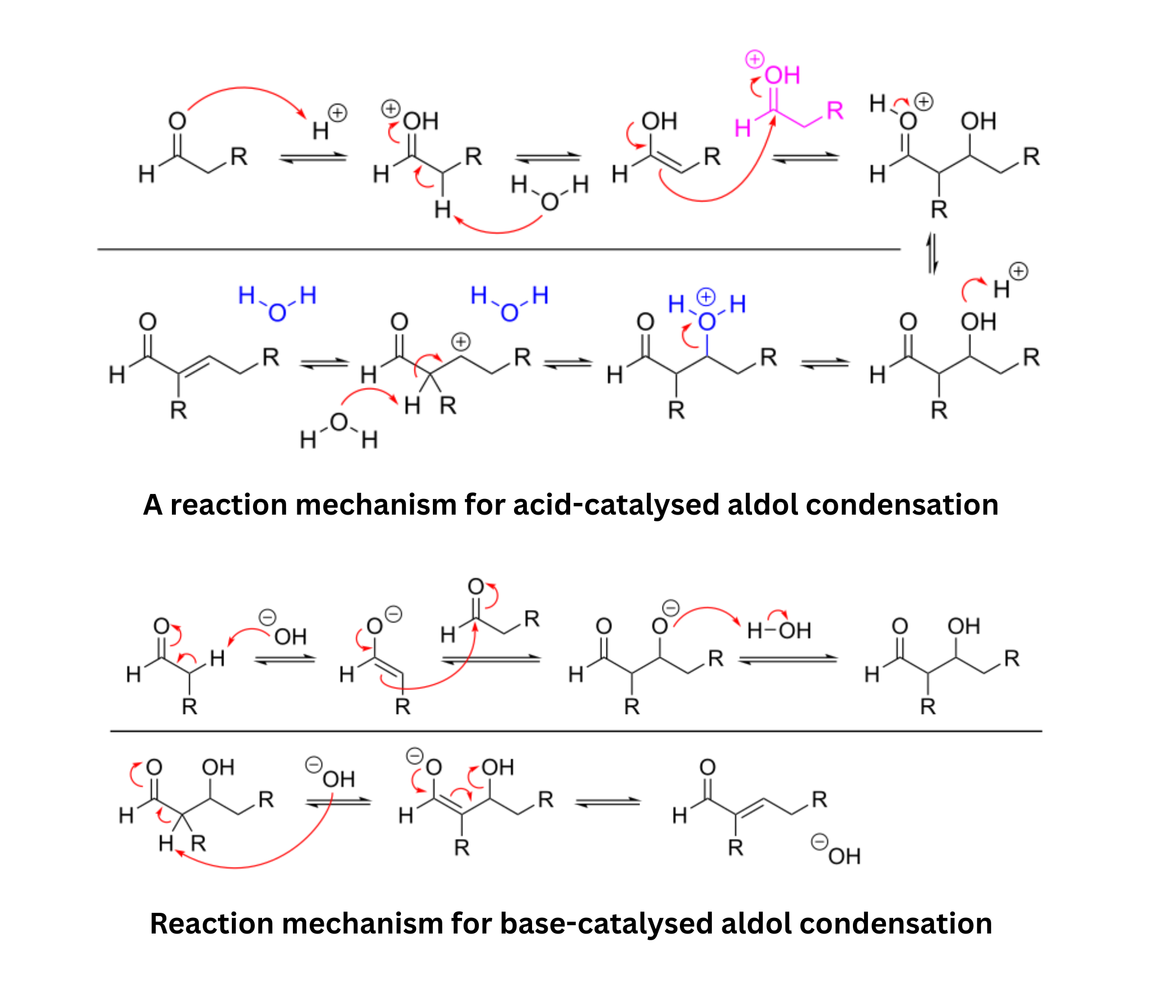
Carbohydrates, aldehydes and ketones
Carbohydrates
Carbohydrates are essential biomolecules in living organisms. They mainly serve as energy sources and structural components. Carbohydrates range from monosaccharides (single sugar units) to polysaccharides (long chains of sugars).
Common examples include glucose, a major cellular fuel, and fructose, found in fruits and in table sugar.
Nomenclature and classification, common names
- Monosaccharides: Single sugar units such as glucose (), galactose, and fructose.
- Disaccharides: Two monosaccharides linked by a glycosidic bond (e.g., sucrose, lactose, maltose).
- Polysaccharides: Long chains of monosaccharides (e.g., starch, glycogen, cellulose).
- Prefixes:
- deoxy indicates removal of an group, replaced by .
- D/L denotes the absolute configuration relative to D-glyceraldehyde or L-glyceraldehyde.
- / specifies the anomeric configuration in cyclic sugars, based on whether the anomeric group is on the opposite side or the same side as the group.
- All sugars end in -ose (e.g., glucose, fructose, galactose).
Absolute configuration
Monosaccharides often contain multiple chiral centers, but the D/L system uses one specific reference point. In the open-chain form, look at the chiral carbon farthest from the carbonyl group; its configuration determines whether the sugar is D or L.
Cyclic structure and conformations of hexoses
In water, many monosaccharides exist mainly in cyclic form. Cyclization occurs when an internal alcohol group reacts with the carbonyl group to form a cyclic hemiacetal (from an aldose) or a hemiketal (from a ketose).
For six-carbon aldoses (e.g., glucose), a six-membered pyranose ring is common. For ketoses (e.g., fructose), a five-membered furanose ring may form.
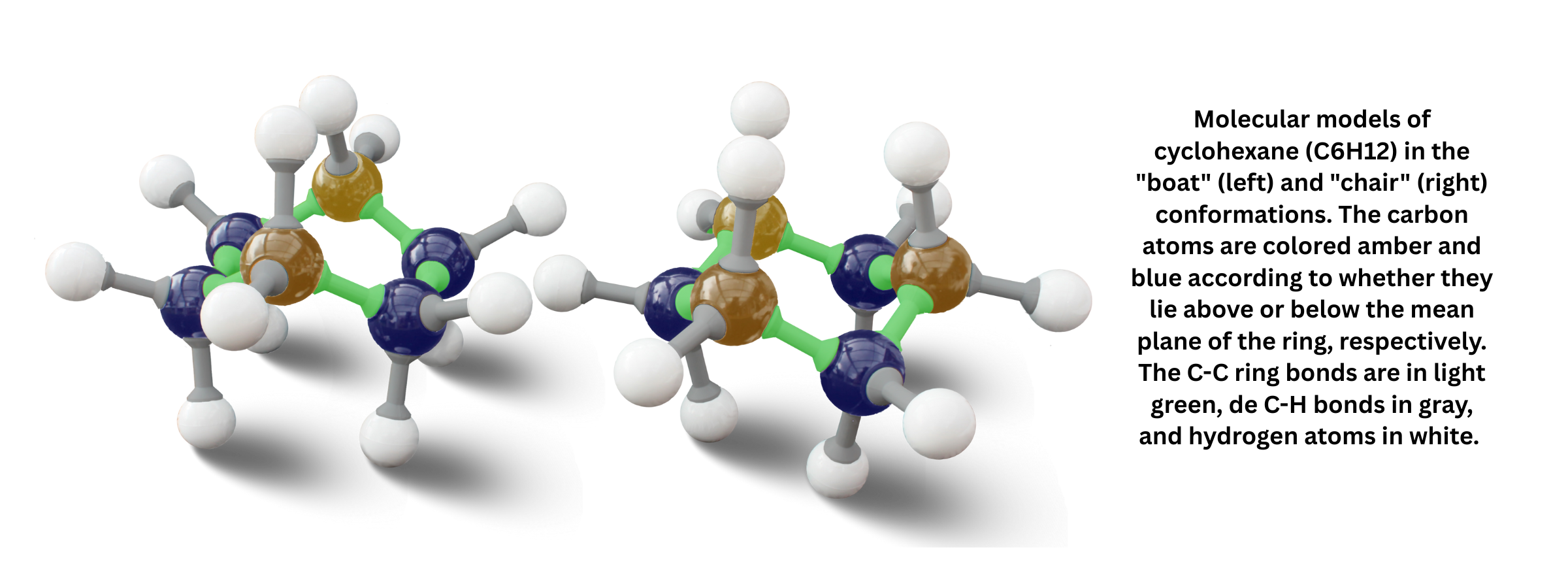
- Chair and boat conformations can occur, with the chair form generally more stable because it minimizes steric hindrance.
Epimers and anomers
- Epimers differ in configuration at one chiral center, excluding the newly formed anomeric center in the ring form.
- Anomers differ specifically at the new chiral center (the anomeric carbon) created during cyclization.
- anomer: The anomeric is oriented opposite the group.
- anomer: The anomeric is oriented on the same side as the group.
Mutarotation is the interconversion between and forms in solution, which changes the observed optical rotation.
Hydrolysis of the glycoside linkage
Glycosidic bonds link monosaccharides to form disaccharides and polysaccharides. Under acidic or enzymatic conditions, these bonds can be hydrolyzed, splitting the carbohydrate into its constituent sugars. In biological systems, enzymes called glycosidases catalyze this process.
Keto-enol tautomerism of monosaccharides
Some monosaccharides (especially those that can adopt an open-chain form with an aldehyde or ketone) can undergo keto-enol tautomerism. In this process, the carbonyl group temporarily converts to an enediol intermediate. Under basic conditions, this can allow an aldose to rearrange into a ketose, or a ketose into an aldose.
This tautomerism is not always prominent at physiological pH, but it helps explain aspects of sugar reactivity and interconversion.
Disaccharides
- Sucrose: Formed by and -fructose linked at their anomeric carbons.
- Lactose: Consists of β-galactose joined to glucose ( or ) through a 1→4 glycosidic bond.
- Maltose: Composed of two glucose units connected via a dehydration reaction, typically linkage.
Polysaccharides
- Starch: Predominantly glycosidic bonds linking glucose; used by plants for energy storage.
- Glycogen: Similar to starch but with more frequent branches, making it a highly branched energy reserve in animals.
- Cellulose (not mentioned above but commonly noted): Features linked glucose units, providing structural support in plant cell walls.
Aldehydes and ketones
Aldehydes and ketones are defined by the carbonyl group (), where carbon is double-bonded to oxygen. In an aldehyde, the carbonyl is at the end of a carbon chain. In a ketone, the carbonyl is within the chain.
These functional groups are important intermediates in organic synthesis because the carbonyl carbon is often reactive and can be converted into many other functional groups.
Nomenclature
Aldehydes typically end in “-al,” as in methanal (formaldehyde) or ethanal (acetaldehyde). Ketones generally end in “-one,” as in propanone (acetone) or butanone.
Common names are widely used, while systematic IUPAC names are based on the longest carbon chain containing the carbonyl group.
Physical properties
Aldehydes and ketones often have higher boiling points than comparable alkanes because the bond is polar. However, because they cannot donate hydrogen bonds, their boiling points are generally lower than those of alcohols.
Many low-molecular-weight aldehydes and ketones are water-soluble because the carbonyl oxygen can accept hydrogen bonds from water.
Important reactions
The carbonyl bond is polarized, leaving the carbonyl carbon partially positive. That electrophilic carbon is a common site for nucleophilic attack, which explains many of the characteristic reactions of aldehydes and ketones.
Nucleophilic addition reactions at bond
In nucleophilic addition, a nucleophile attacks the electrophilic carbonyl carbon. The bond breaks, giving an alkoxide intermediate. Depending on conditions, that intermediate may be protonated or converted into other derivatives.
- Acetal, hemiacetal
When an alcohol reacts with an aldehyde or ketone, a hemiacetal or hemiketal forms first. With excess alcohol under acidic conditions, the hemiacetal can convert into an acetal, where two groups are attached to the same carbon. - Imine, enamine
A primary amine reacts with an aldehyde or ketone to form an imine, where the carbonyl oxygen is replaced by a double-bonded nitrogen. Secondary amines can form an enamine, where an α hydrogen shifts to give a linkage. - Hydride reagents
Reagents such as sodium borohydride () or lithium aluminum hydride () deliver hydride to the carbonyl carbon. This reduces aldehydes to primary alcohols and ketones to secondary alcohols. - Cyanohydrin
Addition of hydrogen cyanide to a carbonyl forms a cyanohydrin, which has both a and an attached to the former carbonyl carbon.
Oxidation of aldehydes
Aldehydes can be oxidized to carboxylic acids under relatively mild conditions. Ketones generally resist oxidation unless conditions are strong enough to break carbon-carbon bonds.
Reactions at adjacent positions: enolate chemistry
An enolate forms when an α hydrogen is removed, producing a resonance-stabilized anion. Enolates are key intermediates in many reactions, including aldol condensation.
- Keto-enol tautomerism (-racemization)
Aldehydes and ketones with an hydrogen can interconvert between keto and enol forms through proton shifts. This equilibrium can lead to racemization at the α carbon if that carbon is chiral. - Aldol condensation, retro-aldol
Enolates can attack other carbonyl compounds to form aldol products. Under some conditions, the aldol product dehydrates to form an , -unsaturated carbonyl. The reverse process, retro-aldol, breaks the bond formed in the aldol reaction and regenerates smaller carbonyl compounds. - Kinetic versus thermodynamic enolate
The kinetic enolate forms faster under conditions that favor the lowest activation energy (often low temperature with a strong, bulky base), typically by removing the most accessible proton. The thermodynamic enolate is more stable and is favored at higher temperatures or with weaker bases, typically giving the more substituted double bond.
General principles
Carbonyl reactivity is largely controlled by the polarity of the bond and by how accessible the carbonyl carbon is to an incoming nucleophile. Substituents can change electron density at the carbonyl carbon, and steric bulk can slow nucleophilic approach.
- Effect of substituents on reactivity of ; steric hindrance
Electron-withdrawing substituents increase the electrophilicity of the carbonyl carbon, making nucleophilic addition more favorable. Bulky substituents hinder approach to the carbonyl center and can reduce reaction rates. - Acidity of -H; carbanions
Hydrogens next to a carbonyl are more acidic than typical alkyl bonds because deprotonation forms a resonance-stabilized carbanion (the enolate). This increased acidity enables many aldol-type and related transformations in synthetic chemistry.