Adrenal disorders
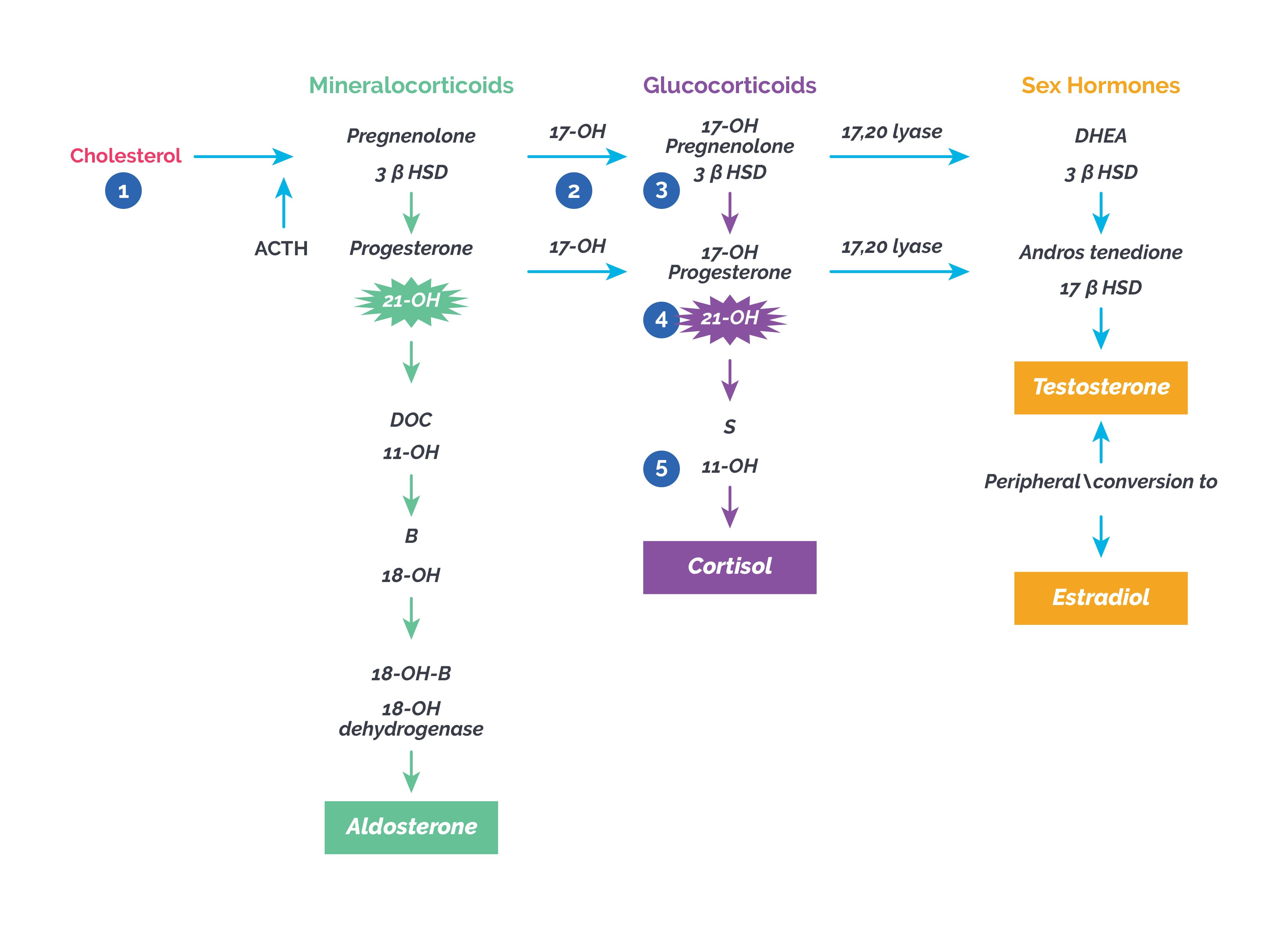
i) Congenital adrenal hyperplasia (CAH): CAH refers to a group of disorders caused by defective adrenal synthesis of steroid hormones. CAH is inherited as an autosomal recessive (AR) disorder. More than 90% of CAH cases are caused by deficiency of the cytochrome P450 enzyme 21-hydroxylase, followed by 11 beta hydroxylase deficiency. Impaired cortisol synthesis leads to chronically elevated ACTH, which causes hyperplasia of the adrenal cortex.
Common features in CAH types
| Disease | Features |
| 21 hydroxylase deficiency | Virilization of female fetus, normal male genitalia; decreased cortisol, increased 17 OH progesterone, DHEA and androstenedione plus salt wasting features due to mineralocorticoid deficiency with high potassium, low sodium and hyperpigmentation seen in salt wasting type; virilizing type has all except salt wasting features; CYP21A gene defect |
| 11 beta hydroxylase deficiency | Females virilized with ambiguous genitalia, males unchanged; low renin HT, elevated DOC, 11-deoxycortisol and androgens, low K, elevated Na; CYP11B1 gene defect |
| 3 beta HSD (hydroxysteroid dehydrogenase deficiency) | Virilization of female fetus, salt wasting, elevated DHEA, pregnenolone and potassium, low androstenedione, testosterone and sodium. In postnatal form, genitalia are normal and salt wasting is absent with mild features of hyperandrogenism |
| 17 alpha hydroxylase deficiency | Low renin hypertension, hypokalemia, metabolic alkalosis, variable sexual development, normal or decreased androgens, estrogens, elevated DOC* and corticosterone |
| 17,20 lyase deficiency | Infantile female genitalia, decreased androgens and estrogens |
*DOC deoxycortisone
Virilization of female external genitalia can present as clitoral enlargement, ambiguous genitalia, and fusion of the labia. In CAH, internal genitalia develop normally.
Patients may also have premature pubarche, acne, infertility, rapid growth, precocious puberty, premature fusion of bony epiphyses, hirsutism, temporal balding, and short adult stature. Some cases may present with primary amenorrhea. PCOS may be associated.
The salt-wasting type is caused by decreased aldosterone synthesis. It presents with poor feeding, weight loss, failure to thrive, vomiting, dehydration, hypotension, hyponatremia, hyperkalemic metabolic acidosis, and potentially fatal adrenal shock. Male newborns are at increased risk because their external genitalia appear normal, so the diagnosis may be missed.
Diagnosis is based on clinical features and confirmed by measuring hormone levels. X-ray shows increased bone age.
Newborn screening for CAH is done by measuring 17-hydroxyprogesterone in blood obtained by heel prick.
The gold standard for diagnosis of 21 hydroxylase deficiency is the corticotropin (ACTH) or cosyntropin stimulation test. This measures 17 hydroxyprogesterone and androstenedione levels at baseline and 1 hour after intravenous cosyntropin. Levels of both hormones are high in 21 hydroxylase deficiency.
Amniotic fluid levels of 17 OH progesterone are also elevated. Chorionic villus sampling at 9-11 weeks of pregnancy can be used for molecular genetic testing for CAH mutations. Dexamethasone administered prenatally helps treat virilization of a female fetus in utero. Fetal DNA can be extracted from maternal blood as early as 4 weeks of gestation, and CAH can be identified by molecular genetic testing.
Postnatal therapy is with glucocorticoids such as hydrocortisone, prednisone, or dexamethasone, which also indirectly suppress excess androgen secretion. Oral contraceptive pills can be used in adult females who develop PCOS. Patients with salt-wasting forms need replacement with fludrocortisone and salt supplementation.