Rate processes in chemical reactions - Kinetics and equilibrium
Reaction rate
A reaction rate describes how quickly reactants are consumed or products form. It’s typically expressed as a change in concentration per unit time, such as molarity per second (M/s). You’ll often see it written in either of these equivalent ways:
The negative sign for a reactant reflects that its concentration decreases as the reaction proceeds.
Dependence of reaction rate on concentration of reactants
The rate law connects the reaction rate to reactant concentrations raised to experimentally determined powers:
The exponents (like , , and ) are determined experimentally by measuring how the rate changes when you change reactant concentrations.
In a multi-step mechanism, these exponents are tied to the rate-determining step (the slow step), not necessarily to the overall balanced equation.
Rate law, rate constant
The rate law’s form (including its exponents) must be determined from experimental data.
The constant is the rate constant. It’s an empirically determined value that depends on reaction conditions (especially temperature).
Reaction order
The overall reaction order is the sum of all exponents in the rate law. A reaction can be:
- Zero-order: The rate does not depend on reactant concentration ( = k).
- First-order: The rate depends on one concentration term to the first power.
- Second-order: The rate depends on either one concentration squared or two separate concentrations multiplied.
- Higher order: Combining multiple reactant concentration terms.
Rate-determining step
In a multi-step reaction, the slowest step is the rate-determining step. This step limits how fast the overall reaction can proceed, so the overall rate is controlled by it.
Because of that, the rate law is typically derived from the reactant concentrations involved in the rate-determining step.
Dependence of reaction rate upon temperature
Reaction rates typically increase as temperature increases. With more thermal energy:
- molecules collide more frequently, and
- a larger fraction of collisions have enough energy to overcome the energy barrier to reach the transition state.
Activation energy
The activation energy () is the minimum energy reactants must have to reach the transition state and form products. Lowering makes it easier for collisions to lead to reaction, which increases the reaction rate.
Activated complex or transition state
At the highest point on an energy profile is the transition state, also called the activated complex. At this point:
- bonds are partially breaking and forming, and
- the system can either fall back to reactants or proceed to products.
Unlike a stable intermediate, the transition state exists only momentarily and cannot be isolated.
Interpretation of energy profiles showing energies of reactants, products, activation energy, and H for the reaction
A reaction energy profile shows how energy changes along the reaction pathway.
- The activation energy is the energy difference from the reactants to the transition state.
- The enthalpy change (H) is the energy difference between products and reactants:
- If H is negative, the reaction is exothermic.
- If H is positive, the reaction is endothermic.
Use of the Arrhenius equation
The Arrhenius equation relates the rate constant to activation energy and temperature:
where is a constant, is the activation energy, is the gas constant, and is the temperature.
This equation shows that increasing or decreasing increases , which increases the reaction rate.
Kinetic control versus thermodynamic control of a reaction
A reaction can produce different products depending on conditions:
- Kinetic product: Forms faster because it has a lower activation energy. It tends to dominate at lower temperatures, where reaction speed matters most.
- Thermodynamic product: More stable (lower free energy) but may require a higher activation energy. It tends to dominate at higher temperatures or when the system has enough time to reach the most stable state.
Catalysts and enzymes lower activation energy, speeding up both forward and reverse reactions without changing the overall thermodynamics. They do not change the position of equilibrium or the G of the process, but they help the system reach equilibrium faster.
Catalysts
A catalyst increases reaction rate by lowering activation energy, but it does not shift the position of equilibrium. It speeds up the forward and reverse reactions equally, so the system reaches the same equilibrium state more quickly.
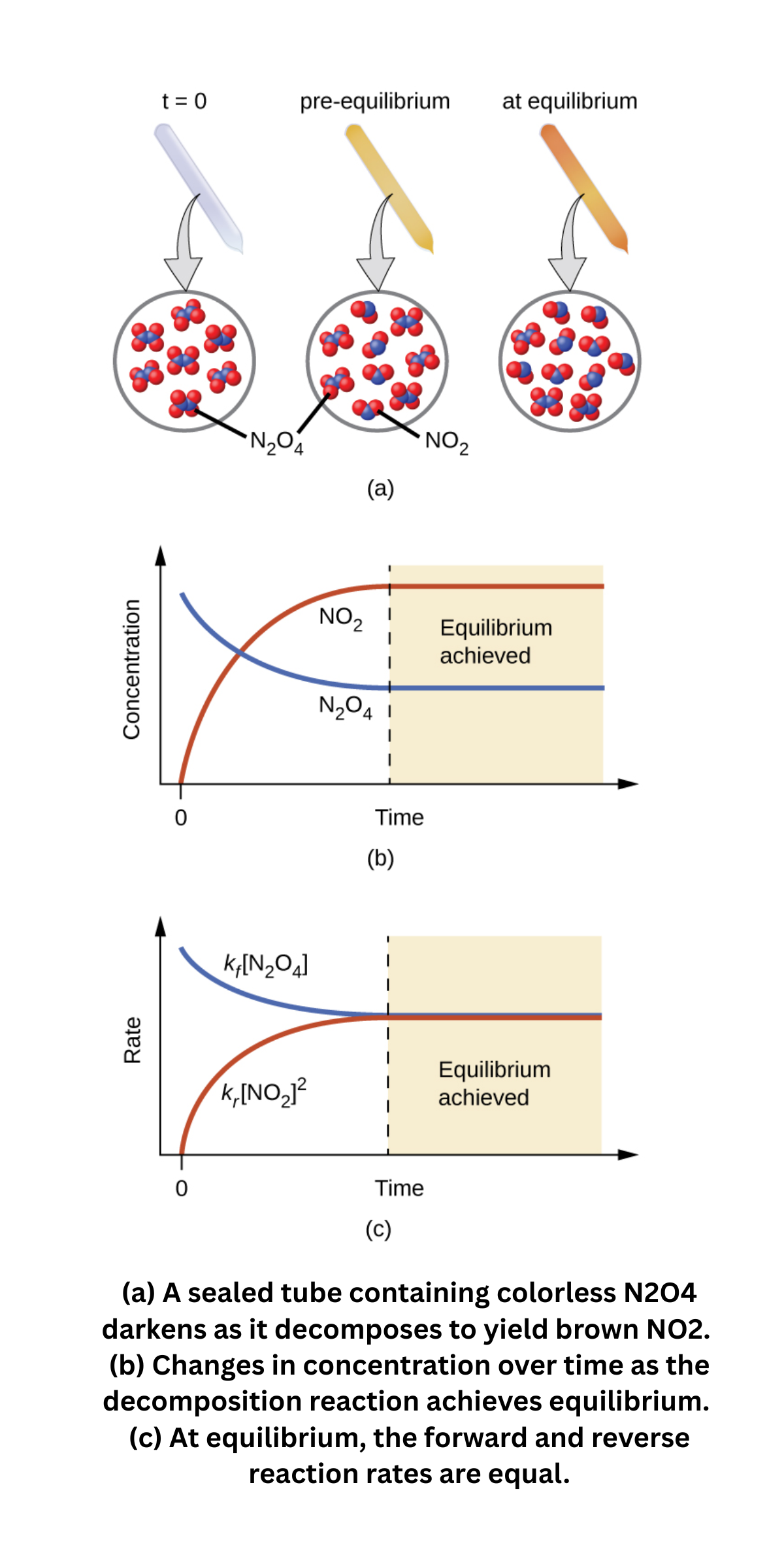
Equilibrium in reversible chemical reactions
Many reactions are reversible: reactants form products, and products can revert to reactants. At equilibrium, the forward and reverse reaction rates are equal.
At that point, concentrations remain constant (though not necessarily equal), even though reactions continue in both directions at the same rate.
Law of mass action
This principle underlies the derivation of equilibrium constants. It states that the rate of a reaction depends on the concentrations (or activities) of the reacting substances. For a generic reaction:
At equilibrium, the forward rate equals the reverse rate:
This leads to the equilibrium constant ():
= .
Equilibrium constant
There are two primary ways to obtain ():
- From the balanced equation, applying the law of mass action.
- From thermodynamics, using the standard free energy relationship:
When (), the reaction lies to the right (favoring products). When (), the reaction balances between reactants and products. When (), the reaction lies to the left (favoring reactants).
Application of Le Châtelier’s principle
If a system at equilibrium is disturbed, it will shift to re-establish equilibrium. Changes in concentration, pressure, volume, or temperature can push the reaction forward or backward.
For example, adding more reactant can shift the system toward products, while adding product can shift it back toward reactants.
Relationship of the equilibrium constant and
Free energy () at any point in a reaction depends on the ratio of products to reactants, expressed by the reaction quotient :
At equilibrium, and , so:
which gives:
Using standard enthalpy and entropy changes to calculate
Use standard enthalpy and entropy data from Appendix G to calculate the standard free energy change for the vaporization of water at room temperature (298 K). What does the computed value for say about the spontaneity of this process?
Solution
The process of interest is the following:
The standard change in free energy may be calculated using the following equation:
From Appendix G:
| Substance | ||
|---|---|---|
Using the appendix data to calculate the standard enthalpy and entropy changes yields:
Substitution into the standard free energy equation yields:
At , so boiling is nonspontaneous (not spontaneous).
Adapted from Example 12.7, OpenStax