Post-streptococcal glomerulonephritis (PSGN)
Post-streptococcal glomerulonephritis (PSGN): PSGN occurs after a streptococcal sore throat or impetigo (skin infection). It’s most common in children ages 3-7 years and is more common in boys. PSGN is a type III hypersensitivity reaction.
It typically presents 1-4 weeks after the streptococcal infection with:
- Edema and periorbital puffiness
- Hematuria (cola-colored urine)
- Hypertension
- Fever and malaise
- Oliguria
BUN and creatinine levels rise. Urinalysis shows RBC casts.
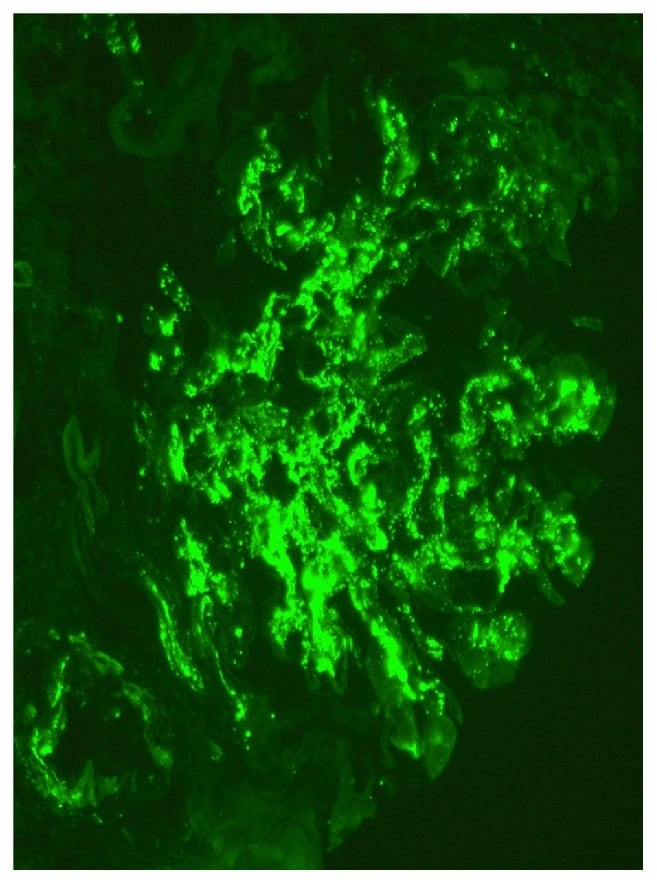
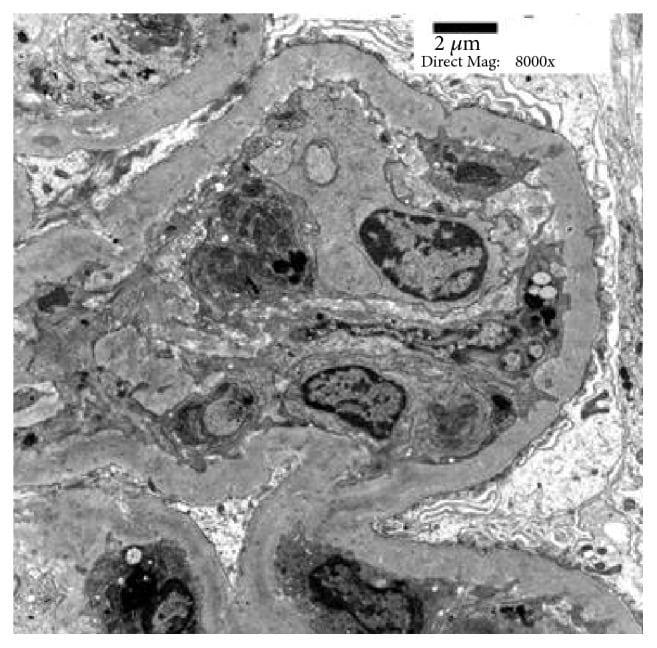
Microscopy shows diffusely enlarged, hypercellular glomeruli due to neutrophil and macrophage infiltration and mesangial proliferation. Immunofluorescence shows lumpy-bumpy (granular) deposits of IgG, IgM, and C3 in the glomeruli. Electron microscopy (EM) shows subepithelial humps composed of immune complex deposits, obliteration of foot processes, and in some cases subendothelial, mesangial, and intramembranous deposits.
Antistreptococcal antibodies are positive. C3 is low for 2-4 weeks and then returns to normal; C4 is normal.
Most cases are transient with full recovery of renal function, but some progress to ESRD. Adults with PSGN have a worse prognosis. Treatment is supportive. Antibiotics may be needed if infection is active, and some cases require steroids and dialysis.
Alport syndrome or hereditary nephritis: Alport syndrome is an inherited disorder transmitted most commonly in an X-linked manner, and less commonly in an autosomal recessive (AR) or autosomal dominant (AD) manner. It’s caused by mutations in the COL4A3, COL4A4, or COL4A5 genes, which code for type IV collagen. (The glomerular basement membrane contains type IV collagen.)
It presents with:
- Progressive loss of renal function
- Microscopic or gross hematuria
- Proteinuria
- Hypertension
- Edema
- Fatigue
- Bilateral sensorineural hearing loss
- Anterior lenticonus
- Vision loss
- Corneal and retinal changes
- Aortic aneurysms
Thin basement membrane disease is a milder form of Alport syndrome without the extra-renal manifestations.
Diagnosis is made with skin or renal biopsy and molecular genetic testing. Immunostaining with antibodies to alpha-5 chains in type IV collagen is negative in skin biopsies, while alpha-5, 3, and 4 chains are present in renal biopsies in Alport syndrome. Electron microscopy shows thinning of the glomerular basement membrane in earlier stages and GBM thickening with alpha 3, 4, and 5 deposits in later stages. Audiometry and an eye exam should be done. Prenatal diagnosis can be done by chorionic villus sampling and amniocentesis.
Treatment includes ACE inhibitors or ARBs to delay progression of renal disease, dialysis, and renal transplant.
IgA nephropathy or Berger’s disease: IgA nephropathy is the most common cause of primary glomerulonephritis. It’s caused by deposition of IgA in the glomerulus and is more common in men.
It typically presents with:
- Gross hematuria
- Flank pain
- Edema and ankle swelling
- Hypertension
Symptoms often follow an upper respiratory tract infection (URTI) or gastrointestinal infection. Some cases progress to end-stage renal disease.
Pathogenesis involves excess amounts of abnormal IgA (poorly galactosylated IgA1), which triggers generation of glycan-specific autoantibodies. IgA immune complexes deposit in the mesangium and activate the alternative complement pathway and possibly the lectin pathway, leading to podocyte damage.
Biopsy shows mesangial proliferation and mesangial deposits. Immunofluorescence shows IgA deposits in the glomerular mesangium, dermis, lung, liver, and intestines.
Mild cases don’t need treatment. When proteinuria exceeds 1 gram/day, ACE inhibitors and ARBs are used. Corticosteroids and cyclophosphamide can be used for severe cases.
Immunofluorescence microscopy showing mesangial deposits of IgA
Lupus nephritis: Lupus nephritis is more common in women and occurs as a complication of SLE. Most cases of lupus are idiopathic, with a possible viral trigger. Drug-induced lupus can occur due to quinidine, methyldopa, chlorpromazine, hydralazine, isoniazid, procainamide, and methyldopa.
Renal pathology in SLE is immune-complex mediated and is considered a type III hypersensitivity reaction. Immune complexes deposit in the mesangium, subendothelial, and subepithelial spaces. There is a lack of immune tolerance to nuclear self-antigens. Nucleic acids released from neutrophils activate intrarenal inflammation, and cytokines such as alpha and beta interferons are released. Complement activation also contributes to renal damage.
WHO classification of lupus nephritis
| Class I | Minimal mesangial , normal microscopy |
| Class II | Mesangioproliferative, mesangial hypercellularity, mesangial immune deposits, minimal capillary wall deposits |
| Class III | Focal, segmental, active (crescentic proliferation) or inactive (sclerosis); <50% glomeruli involved |
| Class IV | Diffuse proliferative glomerulonephritis, >50% glomeruli involved, thick capillary walls with wire-loop lesions, immune deposits of TgG, IgM, IgA, C3, C1q; nephrotic or nephritic syndrome; rapidly progresses to renal failure |
| Class V | Membranous, subepithelial and mesangial immune deposits, spike and dome pattern, nephrotic syndrome |
| Class VI | Advanced sclerotic, >90% glomeruli are globally sclerotic |
It can present with proteinuria, microscopic hematuria, glomerulonephritis, renal failure, and nephritic and nephrotic syndrome. Tubulointerstitial disease and vasculitis can also be seen.
Treatment is a combination of prednisone and cyclophosphamide. Mycophenolate mofetil can be used instead of cyclophosphamide.
Goodpasture syndrome or anti-glomerular basement membrane disease: Goodpasture syndrome is an autoimmune disorder characterized by antibodies against collagen (alpha 3 chain of type IV collagen) in the basement membrane of the alveoli and renal glomeruli. It’s more common in men and can be triggered by cigarette smoking, inhaled hydrocarbons, and viruses.
It has been associated with HLA-DR15 (DR2). Individuals with HLA-DR7 and DR1 have less risk of Goodpasture syndrome. HLA-B8 and DR2 are associated with bad prognosis.
It causes rapidly progressive glomerulonephritis and can present with:
- Hemoptysis
- Edema
- Hematuria
- Proteinuria
- Hypertension
- Cough and dyspnea
- Anemia
- Hepatomegaly
Crackles or rhonchi may be heard on lung auscultation.
Renal biopsy shows focal proliferative to crescentic glomerulonephritis with linear deposits of IgG and complement along the glomerular capillaries. Lung biopsy shows focal necrosis of the alveolar wall, hemosiderin-laden macrophages, and linear deposits of IgG along the alveolar basement membrane.
Treatment includes plasmapheresis to remove antibodies from plasma, steroids, and cyclophosphamide.
Membranoproliferative glomerulonephritis or MPGN: MPGN is a glomerular disorder seen in children and young adults and is characterized by hypocomplementemia. It may present as a nephritic or nephrotic syndrome. Gross hematuria and hypertension are common, and symptoms are often preceded by a respiratory tract infection.
It tends to progress slowly to end-stage renal disease and often recurs after renal transplantation. It may result from cryoglobulinemia, HepB, HepC infections, HIV, bacterial infections like abscess, malaria, leprosy, schistosomiasis, infective endocarditis, SLE, scleroderma, Sjogren’s syndrome, complement deficiency, leukemias, lymphomas, cirrhosis, cystic fibrosis, sarcoidosis, and HUS etc.
Histologically, MPGN is characterized by diffuse mesangial cell proliferation and thickening of capillary walls due to subendothelial extension of the mesangium. Complement activation plays a central role in all forms of MPGN.
There are three types of MPGN as follows:
| Type of MPGN | Features | Biopsy findings |
| Type I (classic) | Most common, activation of classical complement pathway; low or normal C3, low C4 | Diffuse thickening and duplication of GBM, “tram-tracking”, splitting or duplication of the GBM; subendothelial deposits of immune complexes; sclerotic changes |
| Type II (dense deposit disease) | C3 nephritic factor present which causes persistent activation of the alternate complement pathway; associated with macular degeneration and lipodystrophy;worst prognosis; low C3, normal C4 | Eosinophilic deposits within the GBM, “ribbon-like” or “string of sausages” appearance; deposits also in the mesangium, tubular basement membrane and Bowman’s capsule |
| Type III | Low C3, normal C4, low C5-9 | Splitting of GBM, subendothelial and subepithelial deposits |
Treatment includes corticosteroids, cyclophosphamide, cyclosporine, plasma exchange, aspirin, dipyridamole, and rituximab.
Rapidly progressive glomerulonephritis or RPGN or crescentic glomerulonephritis: RPGN is characterized by a rapid decline in renal function. It can present as a nephritic or nephrotic syndrome and may progress to ESRD.
It is seen in Goodpasture syndrome, lupus nephritis, Henoch Schonlein purpura, post-streptococcal glomerulonephritis, ANCA-associated pauci-immune glomerulonephritis (Granulomatosis with polyangiitis (GPA), Churg Strauss syndrome), cryoglobulinemia, and IgA nephropathy.
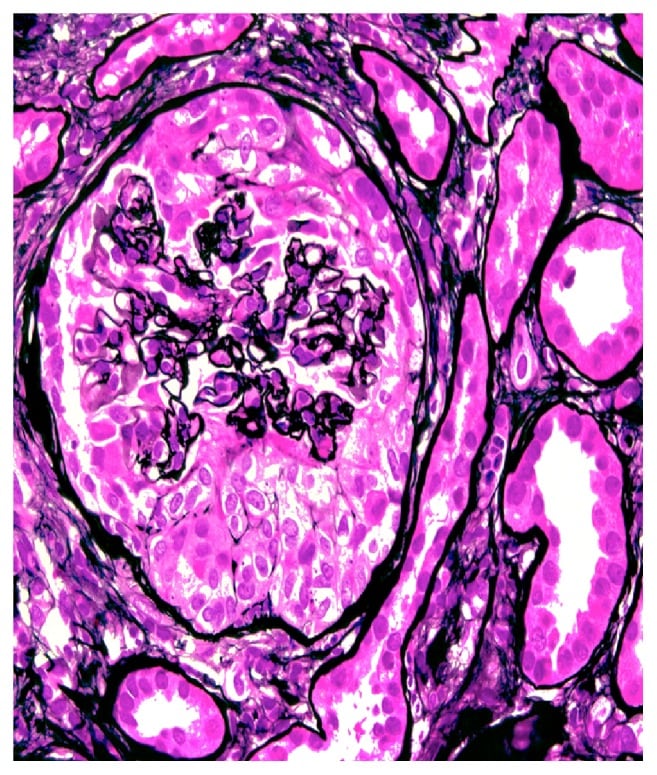
The underlying pathology is severe glomerular injury with rupture of glomerular capillary loops. Biopsy shows characteristic glomerular crescents in >50% of the glomeruli. Crescents result from proliferation of the parietal epithelium of Bowman’s capsule with macrophages, neutrophils, lymphocytes, fibrin, and collagen. Glomerular capillary collapse is seen along with atrophic tubules.
Treatment includes steroids, cyclophosphamide, rituximab, mycophenolate mofetil, plasma exchange, infliximab, etanercept, adalimumab, complement inhibitors, dialysis, and renal transplant, if needed.
Circumferential cellular crescent is present, associated with collapse of the underlying glomerular capillaries (methenamine silver, x400).