Glomerular disorders
- Glomerular disorders: Glomerular diseases include many conditions, with a range of genetic and environmental causes, that interfere with glomerular function. They may present as glomerulonephritis (inflammation of the glomerulus) or glomerulosclerosis (scarring with deposition of hyaline material in the glomerular arterioles and/or mesangium).
Glomerular disease disrupts selective filtration at the glomerulus. As a result, blood components that are normally retained can leak into the urine, leading to hematuria, proteinuria, and loss of other blood components. Clinical presentation depends on the nature and severity of the disease and typically falls into either nephrotic or nephritic syndromes. Some patients have a mixed presentation (e.g., in MPGN).
Nephrotic syndrome: This syndrome is characterized by proteinuria, edema, hypoalbuminemia, hyperlipidemia, and lipiduria. Nephrotic-range proteinuria is > 3.5 gm/24 hours. Urine may appear foamy. Loss of anticoagulants such as antithrombin III leads to a hypercoagulable state and increased platelet aggregation, creating a high risk of venous and arterial thromboembolism. Hyperlipidemia occurs due to increased hepatic synthesis of cholesterol, triglycerides, and lipoproteins; decreased activity of lipoprotein lipase; decreased LDL receptor activity; and increased urinary loss of HDL.
Common causes of nephrotic syndrome
- Minimal change disease (most common cause in children)
- Diabetic nephropathy (most common cause of nephrotic syndrome in adults)
- Focal segmental glomerulosclerosis (second most common cause of nephrotic syndrome in adults)
- Membranous glomerulopathy (third most common cause of nephrotic syndrome in US adults)
- Membranoproliferative glomerulonephritis
- Amyloidosis
Nephritic syndrome: This syndrome is characterized by hematuria, reduced renal function, hypertension, edema, and non-nephrotic proteinuria. Hematuria may appear as cola-colored urine on gross examination.
Common causes of nephritic syndrome
-
Post-streptococcal glomerulonephritis
-
Infective endocarditis
-
IgA nephropathy
-
Lupus nephritis
-
Goodpasture’s syndrome
-
Vasculitis
Minimal change disease (MCD) or lipoid nephrosis: This is the most common cause of nephrotic syndrome in children, though it is also seen in adults. Symptoms are typically preceded by an upper respiratory infection or recent medication use. It may be secondary to HIV, lymphomas, or hypersensitivity reactions to drugs (e.g., NSAIDS) and exposures such as bee stings.
On light microscopy, no changes are seen. Microscopic lipid droplets may be seen in urine and tubular cells. Electron microscopy shows effacement of foot processes. The underlying pathology involves abnormal T cell function with cytokine-induced damage to podocytes. There is an increased Th2 response, overexpression of IL13, and decreased regulatory T cell response. B cell response may be increased. Hemopexin, a plasma protein that binds to sialoglycoproteins in podocytes and causes cytoskeletal rearrangement, may lead to fusion and effacement of foot processes.
Patients are at increased risk of infections such as cellulitis, pneumonia, and peritonitis. Treatment is with corticosteroids, ACE inhibitors and ARBs. Recurrent disease or steroid-resistant cases are treated with cyclophosphamide, chlorambucil, rituximab, cyclosporine, tacrolimus, azathioprine, or mycophenolate mofetil. NSAIDS may reduce proteinuria in some cases. A low-salt diet and diuretics may be used to control edema. Prognosis is typically good.
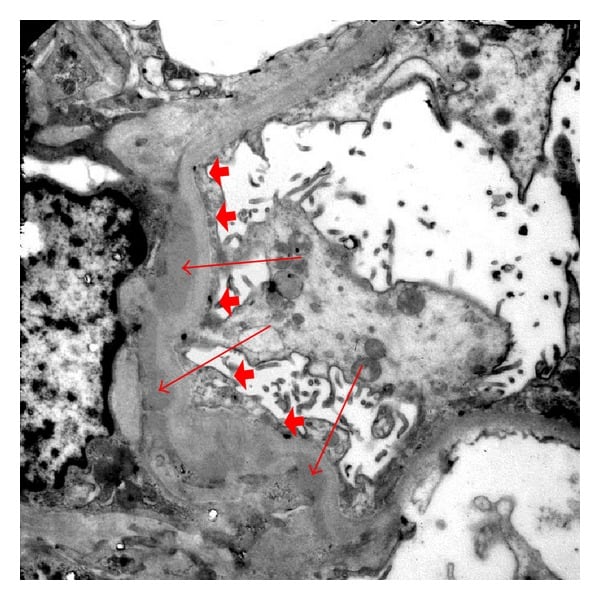
Electron microscopy of renal biopsy with paramesangial deposits (arrows) and foot process effacement (arrow heads).
Focal segmental glomerulosclerosis or FSGS: This disorder is characterized by scarring and hardening (sclerosis) in scattered regions of the kidney. The lesion is typically limited to one part of the glomerulus (segmental) and affects only a minority of glomeruli (focal) in the involved region.
Most cases are idiopathic. FSGS may be secondary to obesity, sickle cell anemia, sleep apnea, diabetes, cyanotic heart disease, unilateral renal agenesis, IUGR, VUR, drugs (e.g., interferons, anthracyclines, calcineurin inhibitors, sirolimus, lithium, bisphosphonates, anabolic steroids), and viruses (e.g., HIV, Hep C, Parvovirus B19, SV 40, EBV, CMV). Some cases are genetic and associated with mutations in the APOL1 gene. FSGS is more common in the African American community.
Podocyte injury is seen in all types of FSGS and may result from cytokines, relative hyperfiltration at the glomerulus, and the effects of interferons following a viral illness. FSGS is a common cause of nephrotic syndrome, and some patients with an aggressive form reach kidney failure in 2 to 3 years. The juxtamedullary (inner cortical) glomeruli are affected first.
Biopsy shows focal and segmental glomerular and mesangial sclerosis, foam cells with lipid inclusions in the capillaries, tubulo-interstitial fibrosis, and hyalinization of the afferent arteriole. Because lesions are focal, multiple areas need to be biopsied. Histologically, the perihilar variant of FSGS has the best prognosis, while the collapsing variant has the worst. IgM and C3 deposits are seen in the sclerotic segments. Electron microscopy shows detachment of epithelial cells from the glomerular basement membrane (GBM) and extensive effacement of podocyte foot processes. The collapsing variant shows collapsed glomerular loops.
Treatment includes steroids, ARBs or ACE inhibitors, immunosuppressive drugs (e.g., cyclosporine, tacrolimus, mycophenolate mofetil), a low-salt and low-protein diet, and diuretics as needed. Severe cases may require dialysis and renal transplantation.
Membranous nephropathy: This is the second most common overall and the most common primary renal disease causing nephrotic syndrome in US adults. Most cases are idiopathic; others are caused by SLE, Hep B, Hep C, some forms of cancer, and drugs/toxins such as penicillamine, gold, mercury, NSAIDS, COX 2 inhibitors, lithium, or captopril. It is more common in caucasians and in those with HLA-DQ1.
It has an autoimmune etiology. Most patients develop antibodies to the M-type phospholipase A2 receptor (anti-PLA2R) and thrombospondin type 1 domain containing 7A (THSD7A). Complement-mediated damage to podocytes may occur.
Biopsy reveals deposits of immunoglobulin G (mainly IgG4) and complement C3, present uniformly in the glomerular capillaries in a subepithelial distribution. Subepithelial “spikes” and GBM thickening may be seen later. The EM appearance of the “spike and dome” pattern is characteristic. Serum anti-PLA2R or THSD7A may be positive.
About 20 percent of patients recover without treatment, while some progress to ESRD (end-stage renal disease). ACE inhibitors and ARBs are generally used to reduce proteinuria. Symptomatic control includes a low-salt and low-protein diet, statins, and diuretics. Resistant cases are treated with steroids, cyclophosphamide, cyclosporine, tacrolimus, rituximab, tetracosactrin (ACTH analogue), or corticotropin. Persistence of anti-PLA2R/THSD7A antibodies is associated with a bad prognosis. Renal transplant may be required.
Diabetic nephropathy (DN): This is the most common cause of nephrotic syndrome in adults and the most common cause of end-stage renal disease. It is more common in males, African Americans, and Mexican Americans. Co-existent hypertension aggravates nephropathy.
Pathophysiology of diabetic nephropathy
Hyperglycemia causes hyperfiltration at the glomerulus and increased glomerular hydrostatic pressure. There is activation of the renin-angiotensin-aldosterone system. Oxidative stress and reactive oxygen species activate cellular pathways such as MAPK, NF-kB, and protein kinase C. Conversion of excess glucose to sorbitol increases the NADH/NAD+ ratio. Hyperglycemia also causes non-enzymatic glycation of proteins, lipids, and nucleic acids, with increased expression of pro-inflammatory cytokines and growth factors such as TGF-beta, PDGF, and VEGF. There is recruitment and activation of T cells and macrophages. Accumulation of interstitial macrophages correlates strongly with proteinuria, interstitial fibrosis, and GFR decline.
DN develops slowly over many years. Asymptomatic microalbuminuria occurs 5-10 years after disease onset. A few years later, this is followed by overt proteinuria of 0.5-3 gm/day and then nephrotic-range proteinuria of > 3.5 gm/day. In type 2 diabetics, more patients have DN at the time of diagnosis because type 2 diabetes can go unrecognized for years. GFR decreases concomitantly.
A spot urine albumin/creatinine ratio ( normal >30 mg/g creatinine) can be used for screening for DN. Serum TNF alpha receptor and uric acid levels correlate with disease prognosis. Biopsy shows mesangial expansion, thickening of the basement membrane, arteriolar hyalinosis, and characteristic nodular glomerulosclerosis (Kimmelstiel-Wilson lesion). Tubular atrophy and interstitial fibrosis are seen. An infiltrate of T cells and macrophages may be present. EM shows podocyte loss and decreased endothelial cell fenestration.
Management involves blood sugar control and control of hypertension. ACE inhibitors and ARBs are renoprotective. Pioglitazone, rosiglitazone, and sitagliptin have beneficial effects on proteinuria and glomerular function. Aldosterone antagonists such as spironolactone (additional anti-inflammatory effect), verapamil and diltiazem (reduce proteinuria), and thiazides or loop diuretics (increase efficacy of ACEI and ARBs) may be used.
Supportive treatment includes Vit D supplementation, keeping cholesterol levels in the normal range, maintaining ideal body weight, aerobic exercise, and a low-salt and low-protein diet. Gene therapy, direct renin inhibitors such as aliskiren, endothelin inhibitors such as atrasentan, PKC inhibitors such as ruboxistaurin, and phosphodiesterase inhibitors such as cilostazol and pentoxifylline are some of the novel therapeutic agents for diabetic nephropathy. Renal transplantation and dialysis are needed once ESRD develops.