Cysts, benign and malignant growths of the urinary system
Polycystic kidney disease (ADPKD and ARPKD): This is an inherited condition in which multiple cysts form in the kidneys and sometimes in other organs (such as the liver). It can be inherited as either autosomal dominant (AD) or autosomal recessive (AR). It’s fairly common: the autosomal dominant form affects about 1 in 500 to 1,000 people, while the autosomal recessive type occurs in an estimated 1 in 20,000 to 40,000 people.
ADPKD is caused by mutations in the PKD1 or PKD2 genes. Cysts begin as dilated collecting tubules and gradually enlarge, causing pressure necrosis of the adjoining renal parenchyma. ADPKD typically presents in the 3rd or 4th decade of life with hypertension, flank pain, hematuria, UTI, nephrolithiasis, and ESRD. Berry aneurysms are seen in some cases. Liver and pancreatic cysts may be present. Cysts involving the arachnoid membrane may cause subdural hematoma, while those affecting the seminal vesicles may cause infertility. MVP, LVH, and aortic root dilation with aortic insufficiency and aortic dissection may occur.
Types of polycystic kidney disease
| Type | Mutated gene | Features |
| ADPKD type 1 | PKD 1, 16 p, polycystin 1 | Most common, thousands of cysts seen beginning in childhood, presents in adulthood |
| ADPKD type 2 | PKD 2, 4 q, polycystin 2 | Thousands of cysts seen beginning in childhood, less severe, presents in adulthood |
| ARPKD (infantile) | PKHD 1, codes for fibrocystin or polyductin | Rarer, presents in-utero or in early childhood, cysts in the kidney and liver, congenital hepatic fibrosis, hypertension, flank pain, oligohydramnios, enlarged kidneys, renal failure, portal hypertension, leukopenia, anemia, splenomegaly |
Acquired polycystic kidney disease is seen in patients on long-term dialysis.
Ultrasound, MRI, and CT are used for diagnosis. Molecular genetic testing can be done to identify gene mutations. Prenatal ultrasound can detect cases in-utero.
Treatment is supportive, including ARBs or ACE inhibitors for HT, pain relief, avoidance of coffee, and management of complications such as cyst hemorrhage or infections. Dialysis and renal transplant are needed once ESRD develops.
Wilms tumor or nephroblastoma or embryoma of the kidney: This is a malignant renal tumor that is most common in young children below the age of 5 years. It’s the most common malignant renal tumor in children < 15 years old.
It’s associated with mutations in WT1 or 2, CTNNB1, WTX or AMER1 (X linked dominant) genes. These genes are involved in regulation of cell growth and cell division. Most cases arise from somatic mutations, while inherited syndromes are either AD or X-linked dominant.
Wilms tumor is also associated with genomic imprinting involving two genes on chromosome 11p, IGF2 and H19. Increased activity of IGF2 and loss of function of H19 gene activity leads to Wilms tumor. It can be a component of WAGR (Wilms tumor, aniridia, genitourinary anomalies and mental retardation) and Beckwith-Wiedemann syndrome.
Wilms tumor may be asymptomatic in early stages and presents later with abdominal mass, pain and swelling, fever, anemia, hematuria, nausea, vomiting, constipation, dyspnea, and high blood pressure. Some cases may be bilateral. It may involve the renal vein, and distant metastases are seen to the lungs, liver, peritoneum, and bone.
Most tumors arise from persistent immature, embryonal renal tissue called nephrogenic rests that constitute dysplasias of the developing kidney. Biopsy shows densely packed small blue cells with scant cytoplasm, arranged in diffuse, nodular, or cord-like patterns, with fibroblast-like stroma and primitive glomeruli, smooth muscle, adipose tissue, bone, cartilage, and epithelial elements (triphasic pattern).
Diagnosis is by ultrasound, CT scan, or MRI. Treatment is with nephrectomy. Chemotherapy with vincristine, dactinomycin, doxorubicin, cyclophosphamide, etoposide, and carboplatin is used for more advanced stages. Radiotherapy is added to the treatment regimen in the presence of distant metastases.
Prognosis is good because most tumors are limited to the capsule at diagnosis, although some cases may recur within the first two years of treatment.
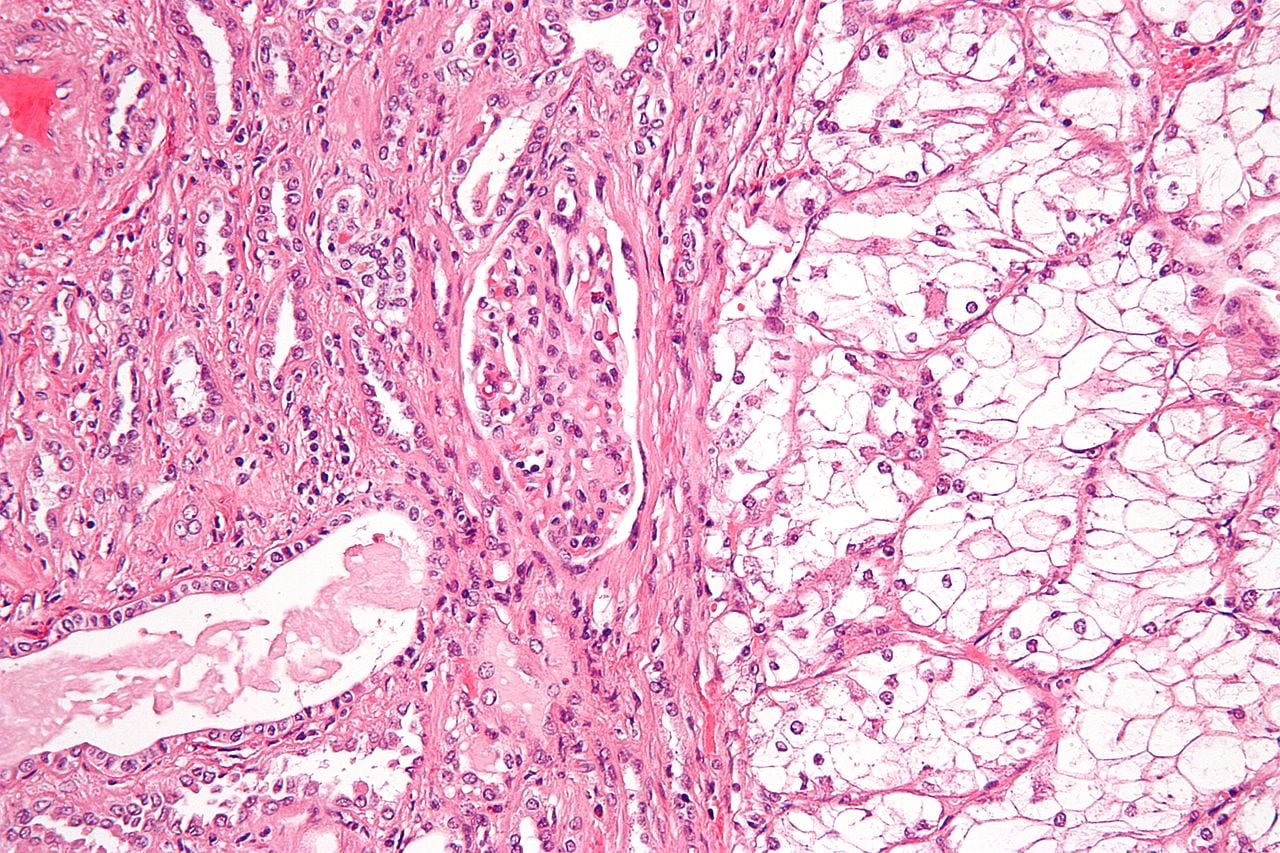
Renal cell carcinoma (RCC) or hypernephroma or Grawitz tumor: This is an adenocarcinoma arising from the tubular epithelium. It’s the most common renal cancer and is more common in men.
Risk factors include smoking and inherited disorders like von Hippel-Lindau disease and tuberous sclerosis, which may show bilateral RCC. Polycystic disease of the kidneys is associated with a higher risk of papillary RCC. Other risk factors include obesity, estrogen therapy, hypertension, analgesic nephropathy, asbestos, heavy metals, and petrochemical exposure.
Mutations in VHL and MET genes are seen. With VHL gene mutation, the VHL protein complex can no longer degrade the transcription factor hypoxia inducible factor (HIF-1 alpha). Uncontrolled activity of HIF leads to increased expression of VEGF, PDGF, and GLUT 1.
It presents with abdominal mass, flank pain, and hematuria, and it’s commonly asymptomatic in early stages. The tumor may extend into the renal vein. Hematogenous metastases are seen to the lung, liver, bone, adrenals, etc.
Microscopic examination shows characteristic cells with clear cytoplasm, arranged in solid, tubular, or trabecular patterns. Clear cells have a lipid- and glycogen-rich cytoplasm, which gets washed off during histopath processing. It shows a characteristic network of thin-walled vessels or “chicken wire” vasculature. Papillary type shows tumor cells forming papilla with fibrovascular stalks.
Treatment is by partial or radical nephrectomy, arterial embolization to debulk tumor mass, external radiation therapy, chemotherapy, interferon, interleukin 2, bevacizumab, kinase inhibitors like anti-VEGF sunitinib and sorafenib, mTOR inhibitors like everolimus, and immunotherapy like Ipilimumab (CTLA 4 inhibitor) and PD 1 inhibitors like nivolumab, pembrolizumab, and avelumab.
High magnification micrograph of a clear cell renal cell carcinoma. H&E stain. Features: Cells with clear cytoplasm, typically arranged in nests. Nuclear atypia is common.
Bladder cancer: The most common bladder malignancies are transitional (urothelial) cell carcinomas, followed by squamous cell carcinomas (more common in Schistosomiasis and squamous metaplasia). Adenocarcinoma is seen in cases of bladder exostrophy.
Risk factors include occupational exposure to benzene and beta naphthyl amines in aniline dyes, rubber, cable, plastic, and textiles industries; bladder infestation with Schistosoma haematobium; leukoplakia of the bladder; obesity; smoking; pelvic radiation; chronic bladder irritation; pioglitazone; and phenacetin-containing analgesic use (banned in North America). It’s more common in men.
It presents with painless gross or microscopic hematuria, increased urgency, frequency of urine, dysuria, nocturia, and incomplete urination or need to strain during urination. Lymph nodes, bone, lung, liver, and peritoneum are the most common sites of metastases.
Bladder cancers appear as papillary or flat, indurated lesions most commonly located in the trigone area, lateral walls, and near the ureteral openings. Biopsy shows either transitional type neoplastic cells or squamous type neoplastic cells with keratin pearls, or rarely adenocarcinoma, which shows gland formation.
Diagnosis is by cystoscopy with biopsy, urine cytology (used for screening as well), CT preferably with urography, MRI, and ultrasound. Renal function tests and urine microscopy for RBCs should also be done. Urine biomarkers for bladder cancer include nuclear matrix protein or NMP 22, CEA, complement factor H related protein, etc.
Cancers that have not involved the muscle layer, including carcinoma-in-situ, are treated with transurethral resection of the tumor followed by intravesical BCG or intravesical chemotherapy. Tumors that have already invaded the muscle are treated with cisplatin-based chemotherapy followed by radical cystectomy and lymphadenectomy. Stage IV and unresectable cancers are treated with chemotherapy.